

Pedigree with frontotemporal lobar degeneration--motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9
- PMID: 18755042
- PMCID: PMC2553097
- DOI: 10.1186/1471-2377-8-32
Pedigree with frontotemporal lobar degeneration--motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9
Abstract
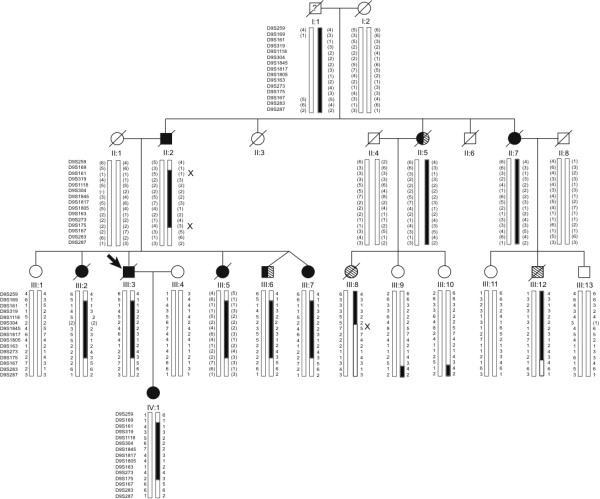
Background: Frontotemporal lobar degeneration (FTLD) represents a clinically, pathologically and genetically heterogenous neurodegenerative disorder, often complicated by neurological signs such as motor neuron-related limb weakness, spasticity and paralysis, parkinsonism and gait disturbances. Linkage to chromosome 9p had been reported for pedigrees with the neurodegenerative disorder, frontotemporal lobar degeneration (FTLD) and motor neuron disease (MND). The objective in this study is to identify the genetic locus in a multi-generational Australian family with FTLD-MND.
Methods: Clinical review and standard neuropathological analysis of brain sections from affected pedigree members. Genome-wide scan using microsatellite markers and single nucleotide polymorphism fine mapping. Examination of candidate genes by direct DNA sequencing.
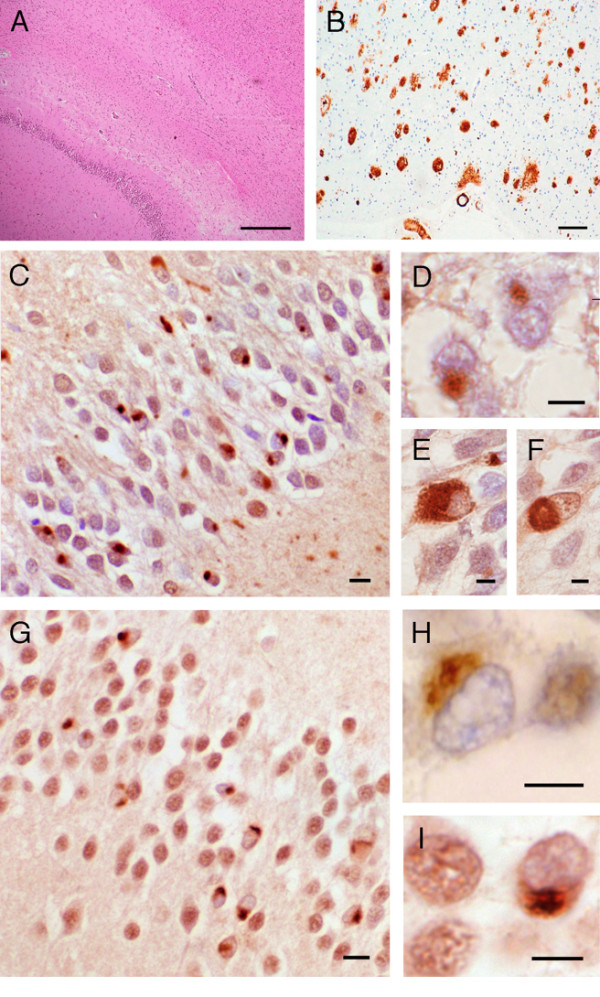
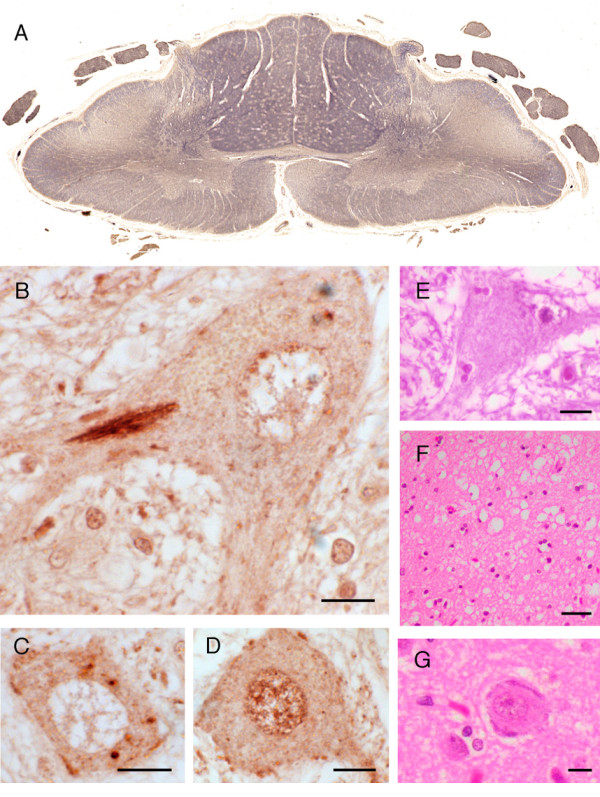
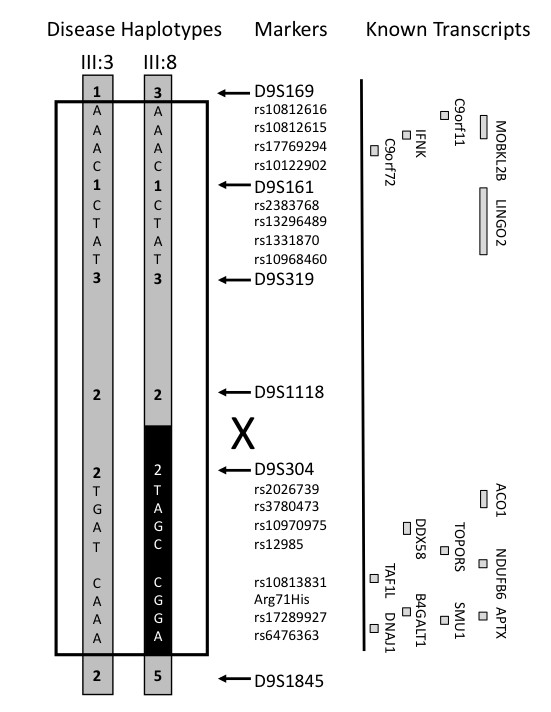
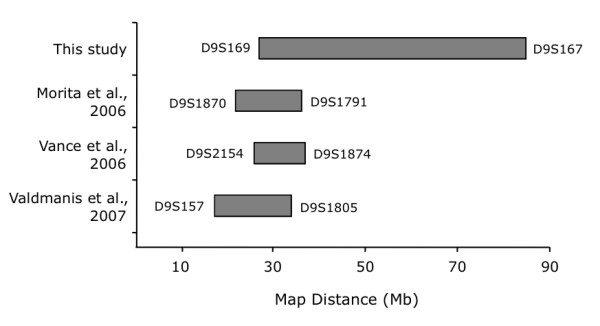
Results: Neuropathological examination revealed cytoplasmic deposition of the TDP-43 protein in three affected individuals. Moreover, we identify a family member with clinical Alzheimer's disease, and FTLD-Ubiquitin neuropathology. Genetic linkage and haplotype analyses, defined a critical region between markers D9S169 and D9S1845 on chromosome 9p21. Screening of all candidate genes within this region did not reveal any novel genetic alterations that co-segregate with disease haplotype, suggesting that one individual carrying a meiotic recombination may represent a phenocopy. Re-analysis of linkage data using the new affection status revealed a maximal two-point LOD score of 3.24 and a multipoint LOD score of 3.41 at marker D9S1817. This provides the highest reported LOD scores from a single FTLD-MND pedigree.
Conclusion: Our reported increase in the minimal disease region should inform other researchers that the chromosome 9 locus may be more telomeric than predicted by published recombination boundaries. Moreover, the existence of a family member with clinical Alzheimer's disease, and who shares the disease haplotype, highlights the possibility that late-onset AD patients in the other linked pedigrees may be mis-classified as sporadic dementia cases.
Figures
References
-
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. - PubMed
-
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. - PubMed
-
- Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL, 3rd, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM, Consortium for Frontotemporal Lobar Degeneration Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol (Berl) 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
