

BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair
- PMID: 18757400
- PMCID: PMC2531069
- DOI: 10.1158/0008-5472.CAN-08-1101
BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair
Abstract
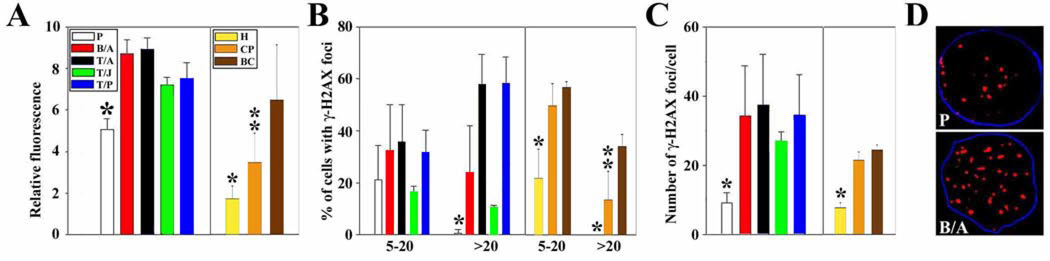
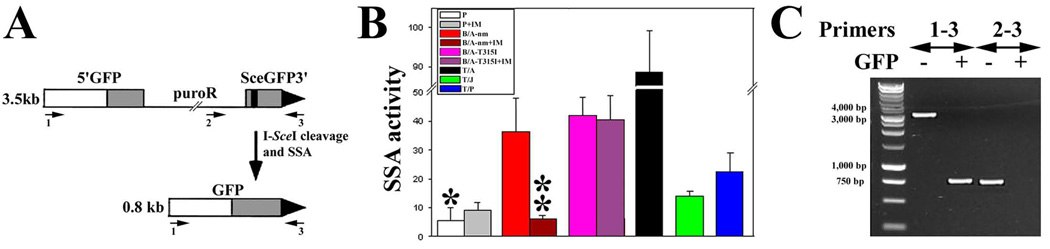
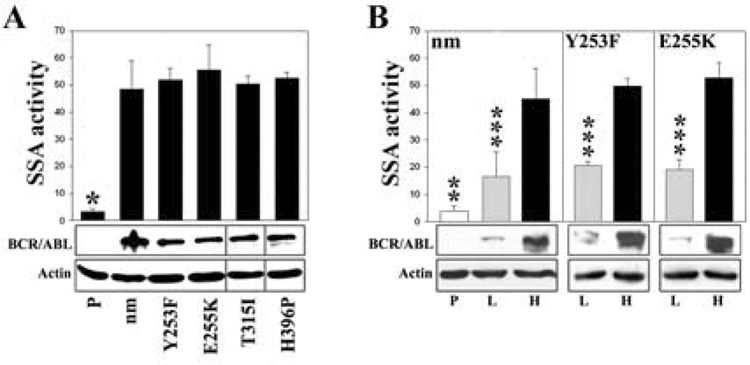
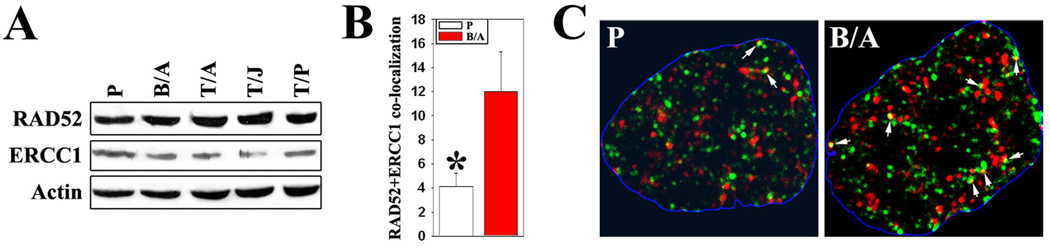
Myeloproliferative disorders (MPD) are stem cell-derived clonal diseases arising as a consequence of acquired aberrations in c-ABL, Janus-activated kinase 2 (JAK2), and platelet-derived growth factor receptor (PDGFR) that generate oncogenic fusion tyrosine kinases (FTK), including BCR/ABL, TEL/ABL, TEL/JAK2, and TEL/PDGFbetaR. Here, we show that FTKs stimulate the formation of reactive oxygen species and DNA double-strand breaks (DSB) both in hematopoietic cell lines and in CD34(+) leukemic stem/progenitor cells from patients with chronic myelogenous leukemia (CML). Single-strand annealing (SSA) represents a relatively rare but very unfaithful DSB repair mechanism causing chromosomal aberrations. Using a specific reporter cassette integrated into genomic DNA, we found that BCR/ABL and other FTKs stimulated SSA activity. Imatinib-mediated inhibition of BCR/ABL abrogated this effect, implicating a kinase-dependent mechanism. Y253F, E255K, T315I, and H396P mutants of BCR/ABL that confer imatinib resistance also stimulated SSA. Increased expression of either nonmutated or mutated BCR/ABL kinase, as is typical of blast phase cells and very primitive chronic phase CML cells, was associated with higher SSA activity. BCR/ABL-mediated stimulation of SSA was accompanied by enhanced nuclear colocalization of RAD52 and ERCC1, which play a key role in the repair. Taken together, these findings suggest a role of FTKs in causing disease progression in MPDs by inducing chromosomal instability through the production of DSBs and stimulation of SSA repair.
Figures
Similar articles
-
BCR/ABL stimulates WRN to promote survival and genomic instability.Cancer Res. 2011 Feb 1;71(3):842-51. doi: 10.1158/0008-5472.CAN-10-1066. Epub 2010 Dec 1. Cancer Res. 2011. PMID: 21123451 Free PMC article.
-
BCR-ABL stimulates mutagenic homologous DNA double-strand break repair via the DNA-end-processing factor CtIP.Carcinogenesis. 2011 Jan;32(1):27-34. doi: 10.1093/carcin/bgq216. Epub 2010 Oct 25. Carcinogenesis. 2011. PMID: 20974687
-
Enhanced phosphorylation of Nbs1, a member of DNA repair/checkpoint complex Mre11-RAD50-Nbs1, can be targeted to increase the efficacy of imatinib mesylate against BCR/ABL-positive leukemia cells.Blood. 2007 Jul 15;110(2):651-60. doi: 10.1182/blood-2006-08-042630. Epub 2007 Apr 12. Blood. 2007. PMID: 17431132 Free PMC article.
-
BCR/ABL, DNA damage and DNA repair: implications for new treatment concepts.Leuk Lymphoma. 2008 Apr;49(4):610-4. doi: 10.1080/03093640701859089. Leuk Lymphoma. 2008. PMID: 18398719 Review.
-
Genomic instability: The cause and effect of BCR/ABL tyrosine kinase.Curr Hematol Malig Rep. 2007 May;2(2):69-74. doi: 10.1007/s11899-007-0010-6. Curr Hematol Malig Rep. 2007. PMID: 20425353 Review.
Cited by
-
Mesenchymal stromal cells in myeloid malignancies: Immunotherapeutic opportunities.Heliyon. 2024 Jan 22;10(3):e25081. doi: 10.1016/j.heliyon.2024.e25081. eCollection 2024 Feb 15. Heliyon. 2024. PMID: 38314300 Free PMC article. Review.
-
Targeting RAD51 phosphotyrosine-315 to prevent unfaithful recombination repair in BCR-ABL1 leukemia.Blood. 2011 Jul 28;118(4):1062-8. doi: 10.1182/blood-2010-09-307256. Epub 2011 Jun 7. Blood. 2011. PMID: 21653319 Free PMC article.
-
Role and mechanism of decitabine combined with tyrosine kinase inhibitors in advanced chronic myeloid leukemia cells.Oncol Lett. 2017 Aug;14(2):1295-1302. doi: 10.3892/ol.2017.6318. Epub 2017 Jun 6. Oncol Lett. 2017. PMID: 28789344 Free PMC article.
-
The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML.Nat Rev Cancer. 2020 Mar;20(3):158-173. doi: 10.1038/s41568-019-0230-9. Epub 2020 Jan 6. Nat Rev Cancer. 2020. PMID: 31907378 Review.
-
BCR-ABL: a multi-faceted promoter of DNA mutation in chronic myelogeneous leukemia.Leukemia. 2010 Jun;24(6):1105-12. doi: 10.1038/leu.2010.67. Epub 2010 May 6. Leukemia. 2010. PMID: 20445577 Free PMC article. Review.
References
-
- Tefferi A, Gilliland DG. Oncogenes in myeloproliferative disorders. Cell Cycle. 2007;6:550–566. - PubMed
-
- Penserga ET, Skorski T. Fusion tyrosine kinases: a result and cause of genomic instability. Oncogene. 2007;26:11–20. - PubMed
-
- Bacher U, Haferlach T, Hiddemann W, et al. Additional clonal abnormalities in Philadelphia-positive ALL and CML demonstrate a different cytogenetic pattern at diagnosis and follow different pathways at progression. Cancer Genet Cytogenet. 2005;157:53–61. - PubMed
-
- Soverini S, Colarossi S, Gnani A, et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006;12:7374–7379. - PubMed
-
- Neviani P, Santhanam R, Trotta R, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8:355–368. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
