

Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis
- PMID: 18769630
- PMCID: PMC2525695
- DOI: 10.1172/JCI34871
Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis
Abstract
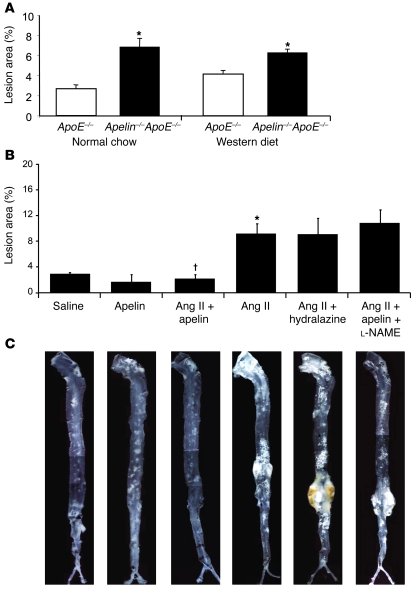
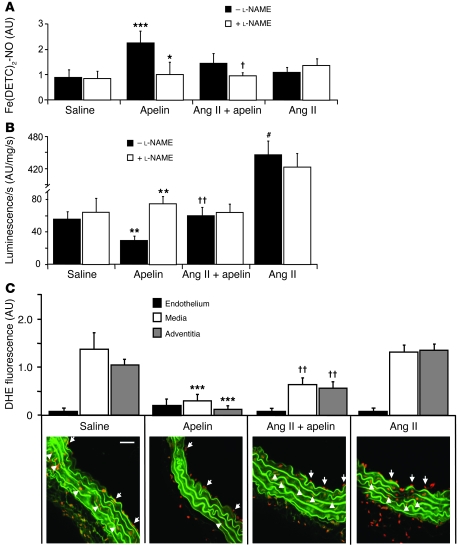
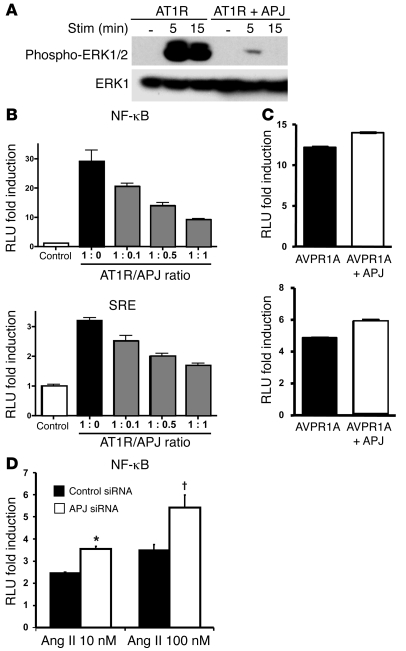
Apelin and its cognate G protein-coupled receptor APJ constitute a signaling pathway with a positive inotropic effect on cardiac function and a vasodepressor function in the systemic circulation. The apelin-APJ pathway appears to have opposing physiological roles to the renin-angiotensin system. Here we investigated whether the apelin-APJ pathway can directly antagonize vascular disease-related Ang II actions. In ApoE-KO mice, exogenous Ang II induced atherosclerosis and abdominal aortic aneurysm formation; we found that coinfusion of apelin abrogated these effects. Similarly, apelin treatment rescued Ang II-mediated increases in neointimal formation and vascular remodeling in a vein graft model. NO has previously been implicated in the vasodepressor function of apelin; we found that apelin treatment increased NO bioavailability in ApoE-KO mice. Furthermore, infusion of an NO synthase inhibitor blocked the apelin-mediated decrease in atherosclerosis and aneurysm formation. In rat primary aortic smooth muscle cells, apelin inhibited Ang II-mediated transcriptional regulation of multiple targets as measured by reporter assays. In addition, we demonstrated by coimmunoprecipitation and fluorescence resonance energy transfer analysis that the Ang II and apelin receptors interacted physically. Taken together, these findings indicate that apelin signaling can block Ang II actions in vascular disease by increasing NO production and inhibiting Ang II cellular signaling.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
