

PhyloGibbs-MP: module prediction and discriminative motif-finding by Gibbs sampling
- PMID: 18769735
- PMCID: PMC2518514
- DOI: 10.1371/journal.pcbi.1000156
PhyloGibbs-MP: module prediction and discriminative motif-finding by Gibbs sampling
Abstract
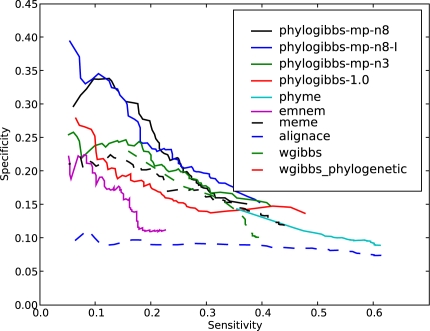
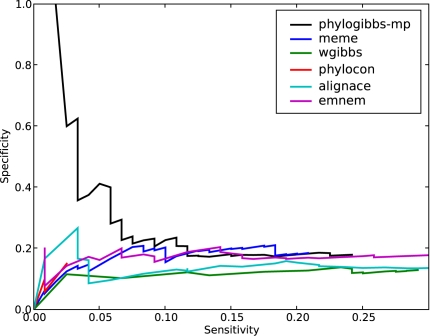
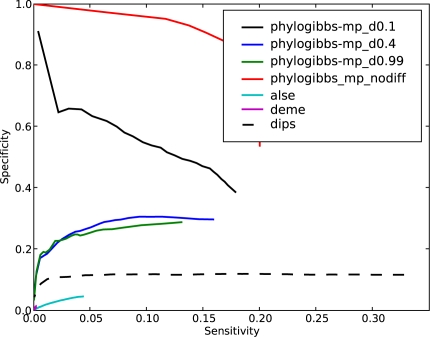
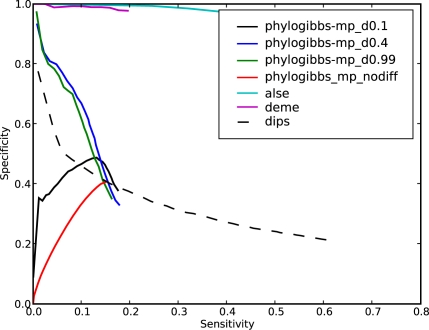
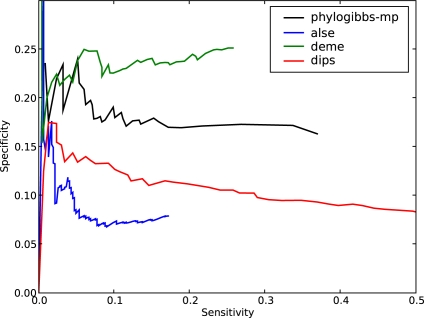
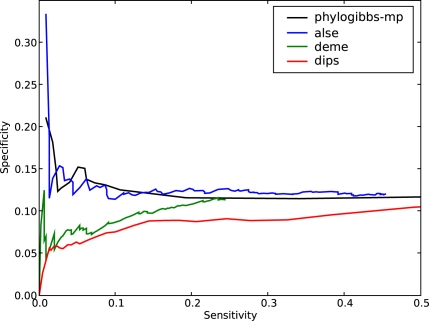
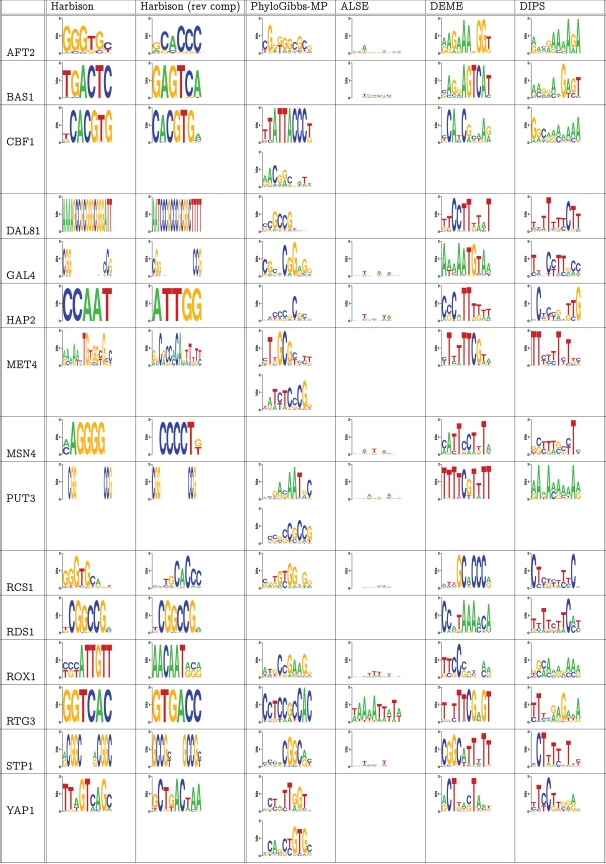
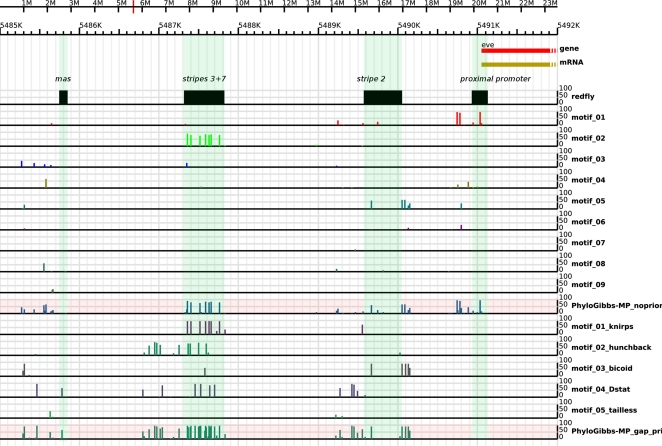
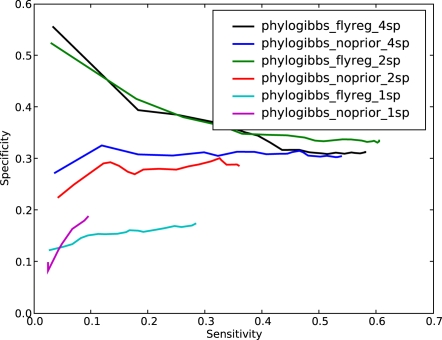
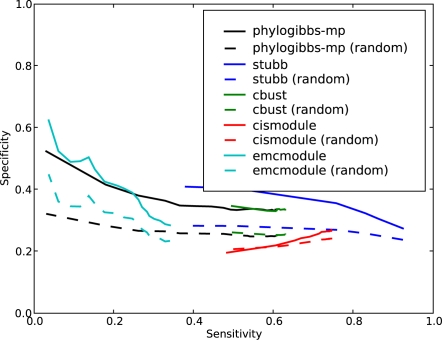
PhyloGibbs, our recent Gibbs-sampling motif-finder, takes phylogeny into account in detecting binding sites for transcription factors in DNA and assigns posterior probabilities to its predictions obtained by sampling the entire configuration space. Here, in an extension called PhyloGibbs-MP, we widen the scope of the program, addressing two major problems in computational regulatory genomics. First, PhyloGibbs-MP can localise predictions to small, undetermined regions of a large input sequence, thus effectively predicting cis-regulatory modules (CRMs) ab initio while simultaneously predicting binding sites in those modules-tasks that are usually done by two separate programs. PhyloGibbs-MP's performance at such ab initio CRM prediction is comparable with or superior to dedicated module-prediction software that use prior knowledge of previously characterised transcription factors. Second, PhyloGibbs-MP can predict motifs that differentiate between two (or more) different groups of regulatory regions, that is, motifs that occur preferentially in one group over the others. While other "discriminative motif-finders" have been published in the literature, PhyloGibbs-MP's implementation has some unique features and flexibility. Benchmarks on synthetic and actual genomic data show that this algorithm is successful at enhancing predictions of differentiating sites and suppressing predictions of common sites and compares with or outperforms other discriminative motif-finders on actual genomic data. Additional enhancements include significant performance and speed improvements, the ability to use "informative priors" on known transcription factors, and the ability to output annotations in a format that can be visualised with the Generic Genome Browser. In stand-alone motif-finding, PhyloGibbs-MP remains competitive, outperforming PhyloGibbs-1.0 and other programs on benchmark data.
Conflict of interest statement
The author has declared that no competing interests exist.
Figures
References
-
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, et al. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. - PubMed
-
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
