

Design of a grid service-based platform for in silico protein-ligand screenings
- PMID: 18771812
- PMCID: PMC2665129
- DOI: 10.1016/j.cmpb.2008.07.005
Design of a grid service-based platform for in silico protein-ligand screenings
Abstract
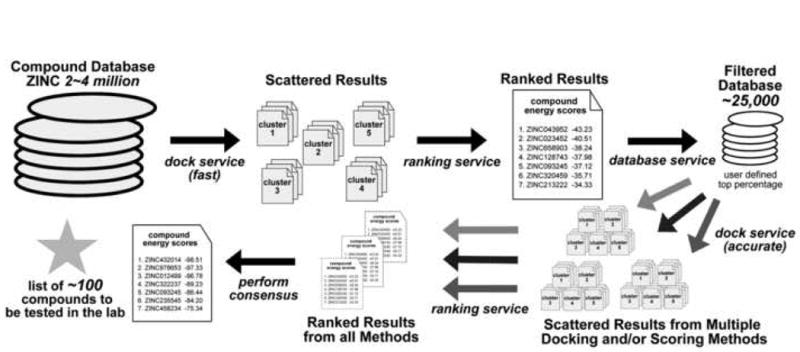
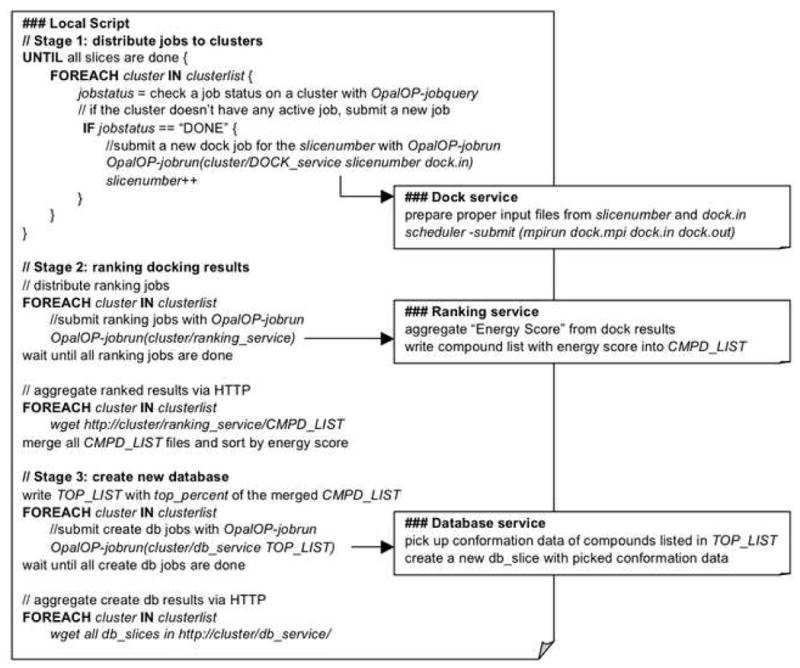
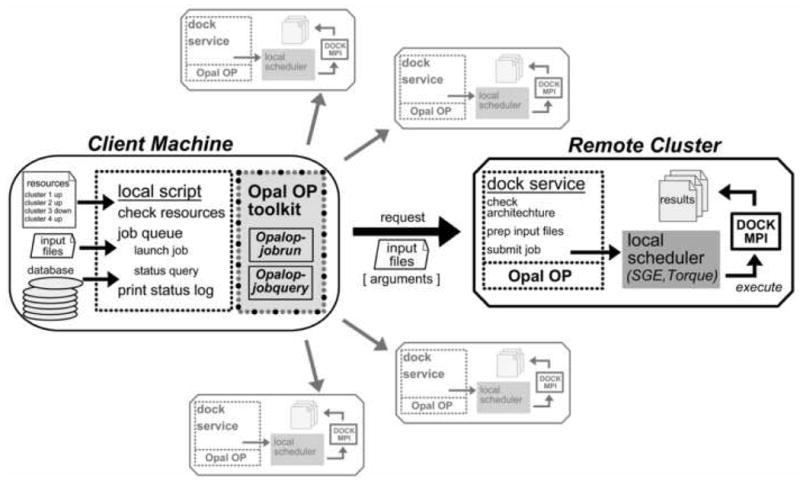
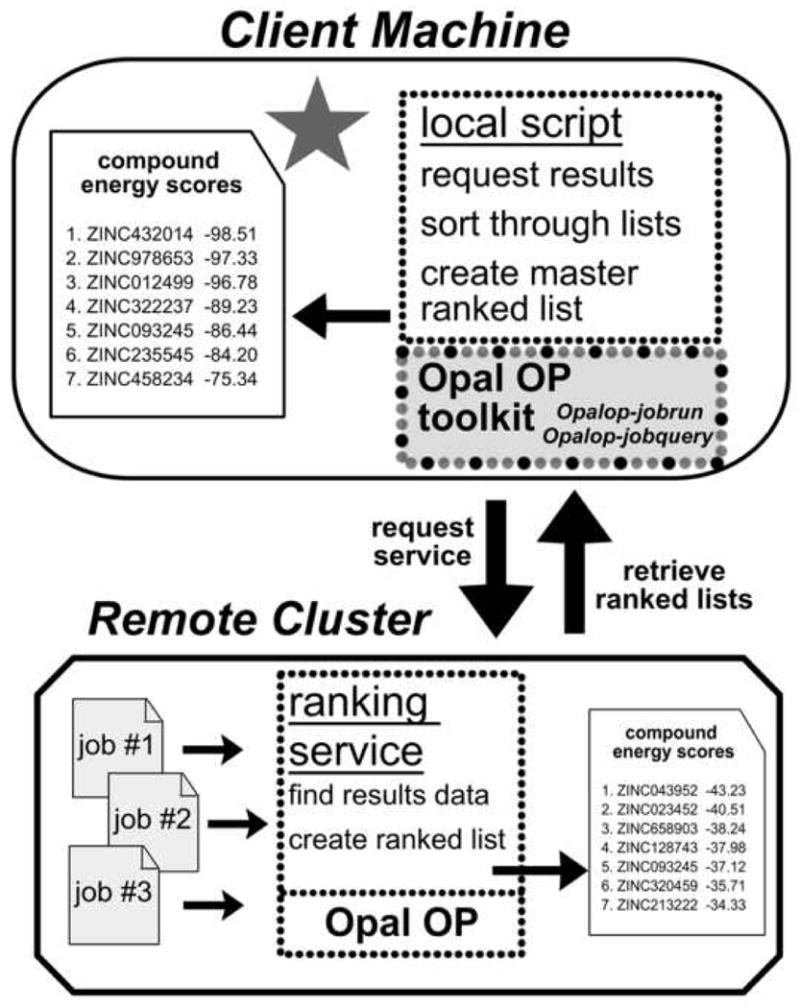
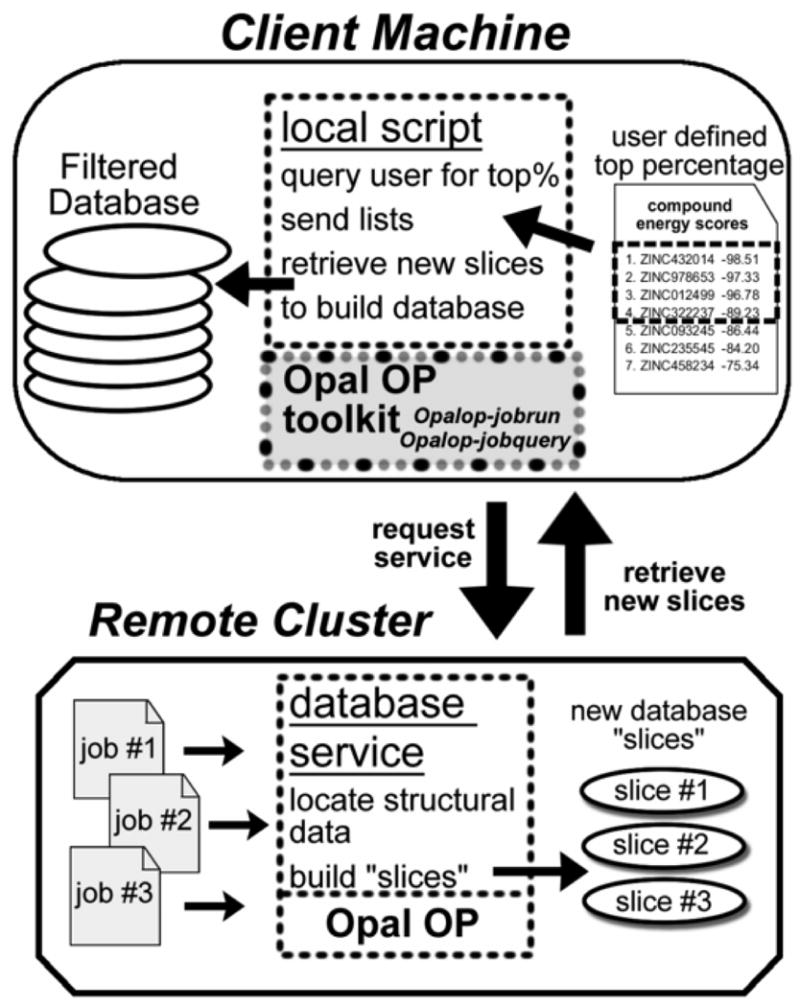
Grid computing offers the powerful alternative of sharing resources on a worldwide scale, across different institutions to run computationally intensive, scientific applications without the need for a centralized supercomputer. Much effort has been put into development of software that deploys legacy applications on a grid-based infrastructure and efficiently uses available resources. One field that can benefit greatly from the use of grid resources is that of drug discovery since molecular docking simulations are an integral part of the discovery process. In this paper, we present a scalable, reusable platform to choreograph large virtual screening experiments over a computational grid using the molecular docking simulation software DOCK. Software components are applied on multiple levels to create automated workflows consisting of input data delivery, job scheduling, status query, and collection of output to be displayed in a manageable fashion for further analysis. This was achieved using Opal OP to wrap the DOCK application as a grid service and PERL for data manipulation purposes, alleviating the requirement for extensive knowledge of grid infrastructure. With the platform in place, a screening of the ZINC 2,066,906 compound "drug-like" subset database against an enzyme's catalytic site was successfully performed using the MPI version of DOCK 5.4 on the PRAGMA grid testbed. The screening required 11.56 days laboratory time and utilized 200 processors over 7 clusters.
Conflict of interest statement
Figures
References
-
- Foster I, Kesselman C, Tuecke S. The anatomy of the grid: Enabling scalable virtual organization. J-IJHPCA. 2001;15(3):200–222.
-
- W3C Web Services Architechture. 2004. http://www.w3.org/TR/ws-arch/.
-
- Foster I, Kesselman C, Nick JM, et al. In: The physiology of the grid, in Grid Computing. Berman Fran, Fox Geoffrey, Hey Tony., editors. John Wiley & Sons, Ltd; West Sussex, England: 2003. pp. 217–249.
-
- Krishnan S, Stearn B, Bhatia K, et al. Opal: Simple web services wrappers for scientific applications. ICWS. 2006:823–832.
-
- Stevens R. Trends in cyberinfrastructure for bioinformatics and computational biology. CTWatch Quarterly. 2006. pp. 1–5. Available: http://www.ctwatch.org/quarterly.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
