

A beta-lactam antibiotic dampens excitotoxic inflammatory CNS damage in a mouse model of multiple sclerosis
- PMID: 18773080
- PMCID: PMC2522272
- DOI: 10.1371/journal.pone.0003149
A beta-lactam antibiotic dampens excitotoxic inflammatory CNS damage in a mouse model of multiple sclerosis
Erratum in
- PLoS ONE. 2008;3(9). doi: 10.1371/annotation/b898d6ee-801a-474e-9776-72e4da664b89
- PLoS One. 2011;6(1). doi: 10.1371/annotation/b1ff1c14-fa84-40a0-b095-e5ee47c74125
Abstract
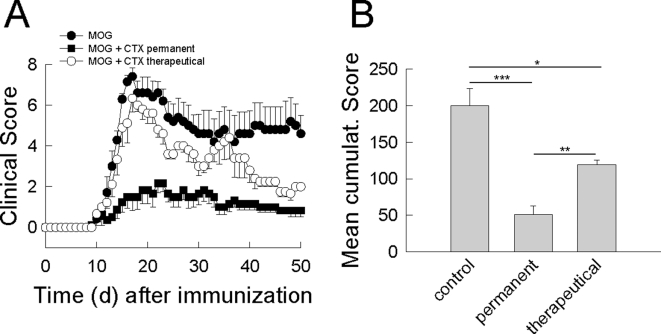
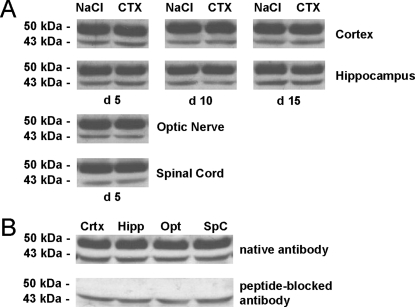
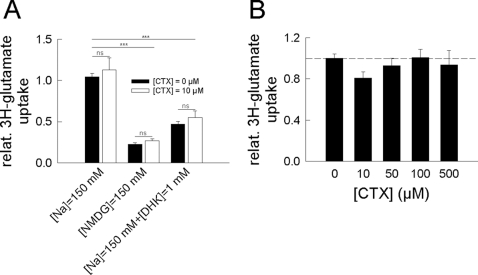
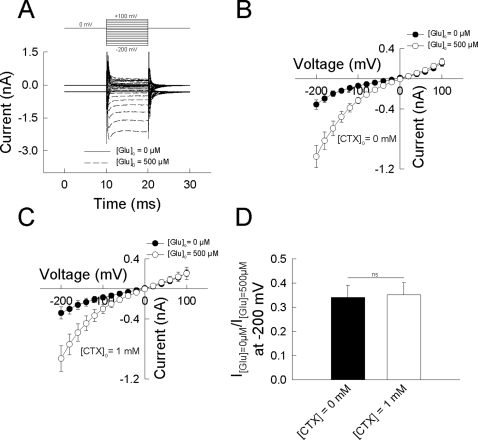
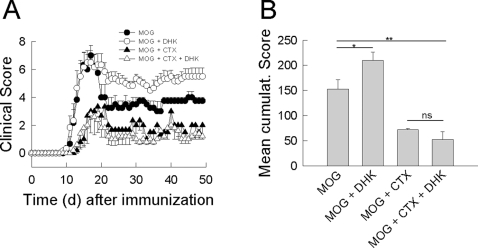
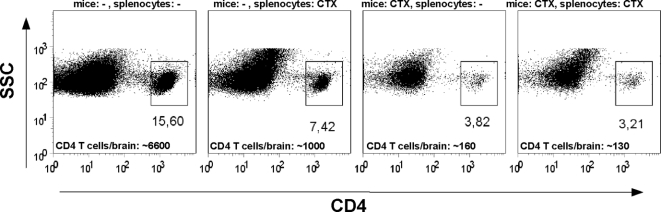
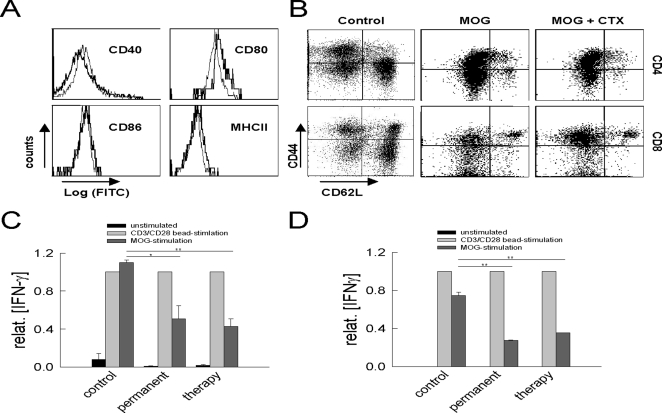
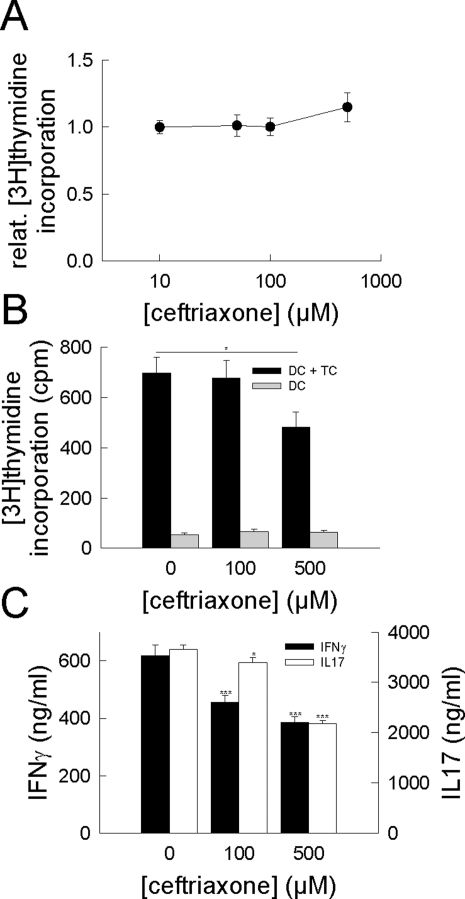
In multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE), impairment of glial "Excitatory Amino Acid Transporters" (EAATs) together with an excess glutamate-release by invading immune cells causes excitotoxic damage of the central nervous system (CNS). In order to identify pathways to dampen excitotoxic inflammatory CNS damage, we assessed the effects of a beta-lactam antibiotic, ceftriaxone, reported to enhance expression of glial EAAT2, in "Myelin Oligodendrocyte Glycoprotein" (MOG)-induced EAE. Ceftriaxone profoundly ameliorated the clinical course of murine MOG-induced EAE both under preventive and therapeutic regimens. However, ceftriaxone had impact neither on EAAT2 protein expression levels in several brain areas, nor on the radioactive glutamate uptake capacity in a mixed primary glial cell-culture and the glutamate-induced uptake currents in a mammalian cell line mediated by EAAT2. Moreover, the clinical effect of ceftriaxone was preserved in the presence of the EAAT2-specific transport inhibitor, dihydrokainate, while dihydrokainate alone caused an aggravated EAE course. This demonstrates the need for sufficient glial glutamate uptake upon an excitotoxic autoimmune inflammatory challenge of the CNS and a molecular target of ceftriaxone other than the glutamate transporter. Ceftriaxone treatment indirectly hampered T cell proliferation and proinflammatory INFgamma and IL17 secretion through modulation of myelin-antigen presentation by antigen-presenting cells (APCs) e.g. dendritic cells (DCs) and reduced T cell migration into the CNS in vivo. Taken together, we demonstrate, that a beta-lactam antibiotic attenuates disease course and severity in a model of autoimmune CNS inflammation. The mechanisms are reduction of T cell activation by modulation of cellular antigen-presentation and impairment of antigen-specific T cell migration into the CNS rather than or modulation of central glutamate homeostasis.
Conflict of interest statement
Figures
References
-
- Frohman EM, Racke MK, Raine CS. Multiple sclerosis–the plaque and its pathogenesis. N Engl J Med. 2006;354:942–955. - PubMed
-
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. - PubMed
-
- Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. - PubMed
-
- Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol. 2001;14:271–278. - PubMed
-
- Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;50:169–180. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
