

The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications
- PMID: 18774188
- PMCID: PMC10124615
- DOI: 10.1016/j.tins.2008.07.003
The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications
Abstract
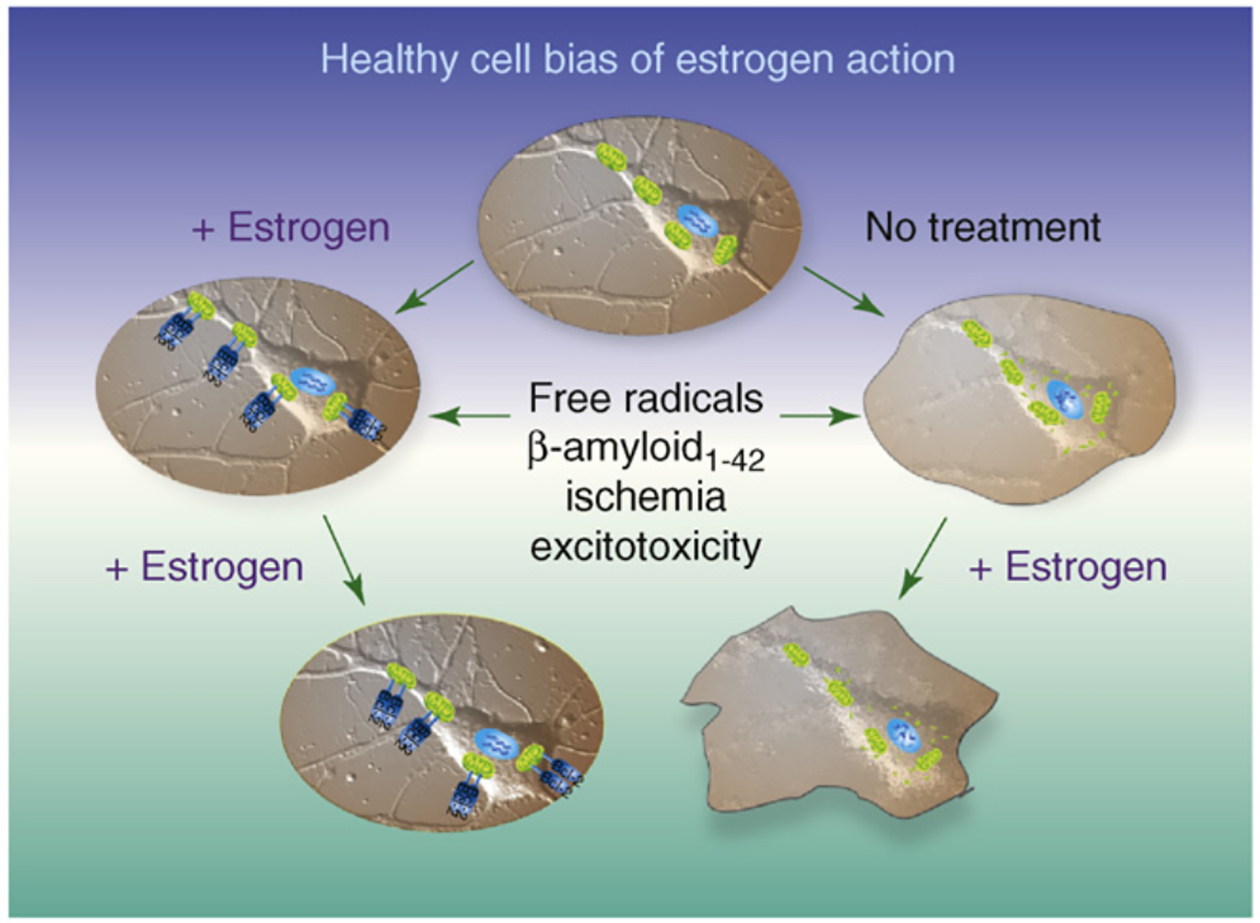
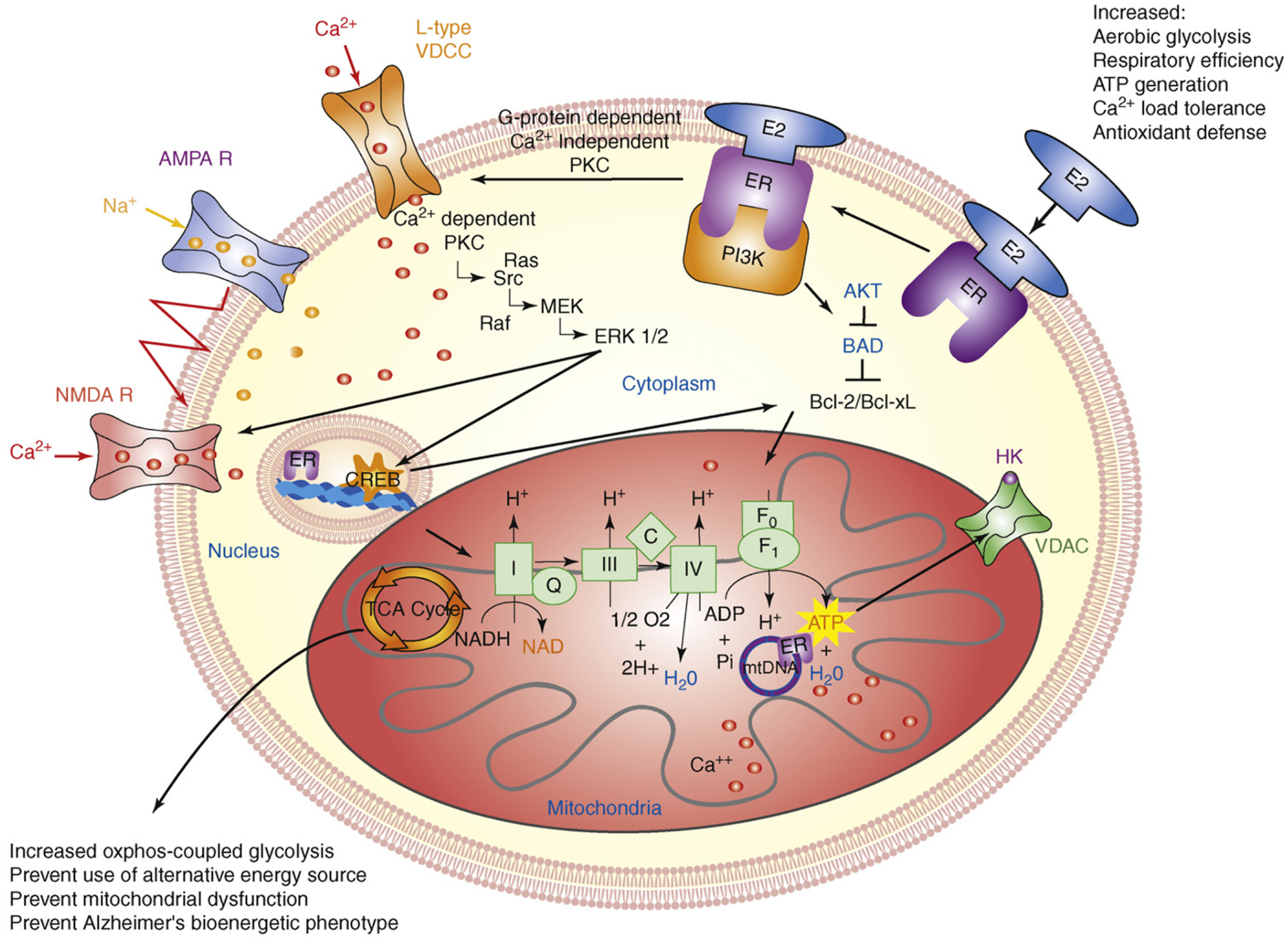
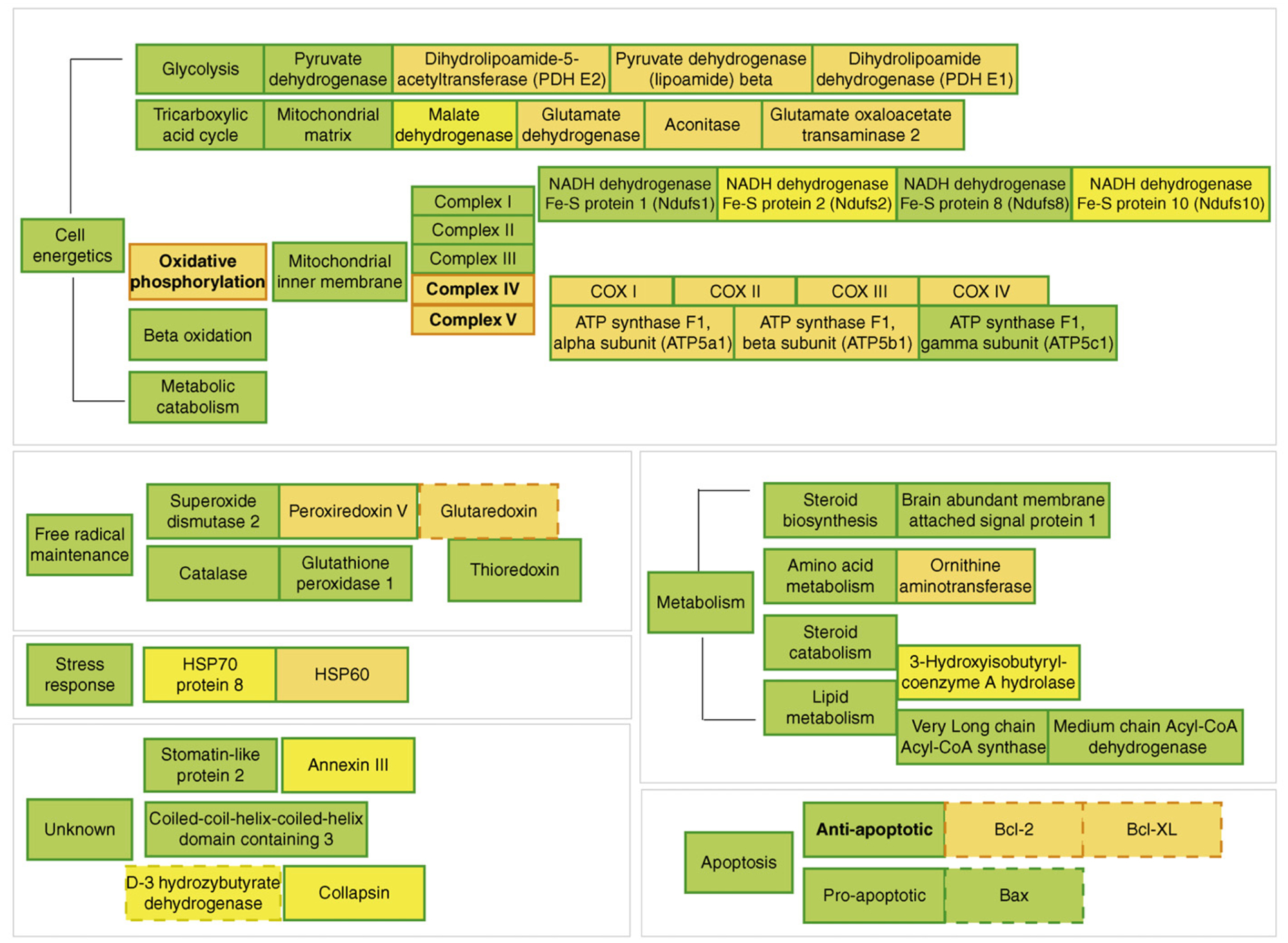
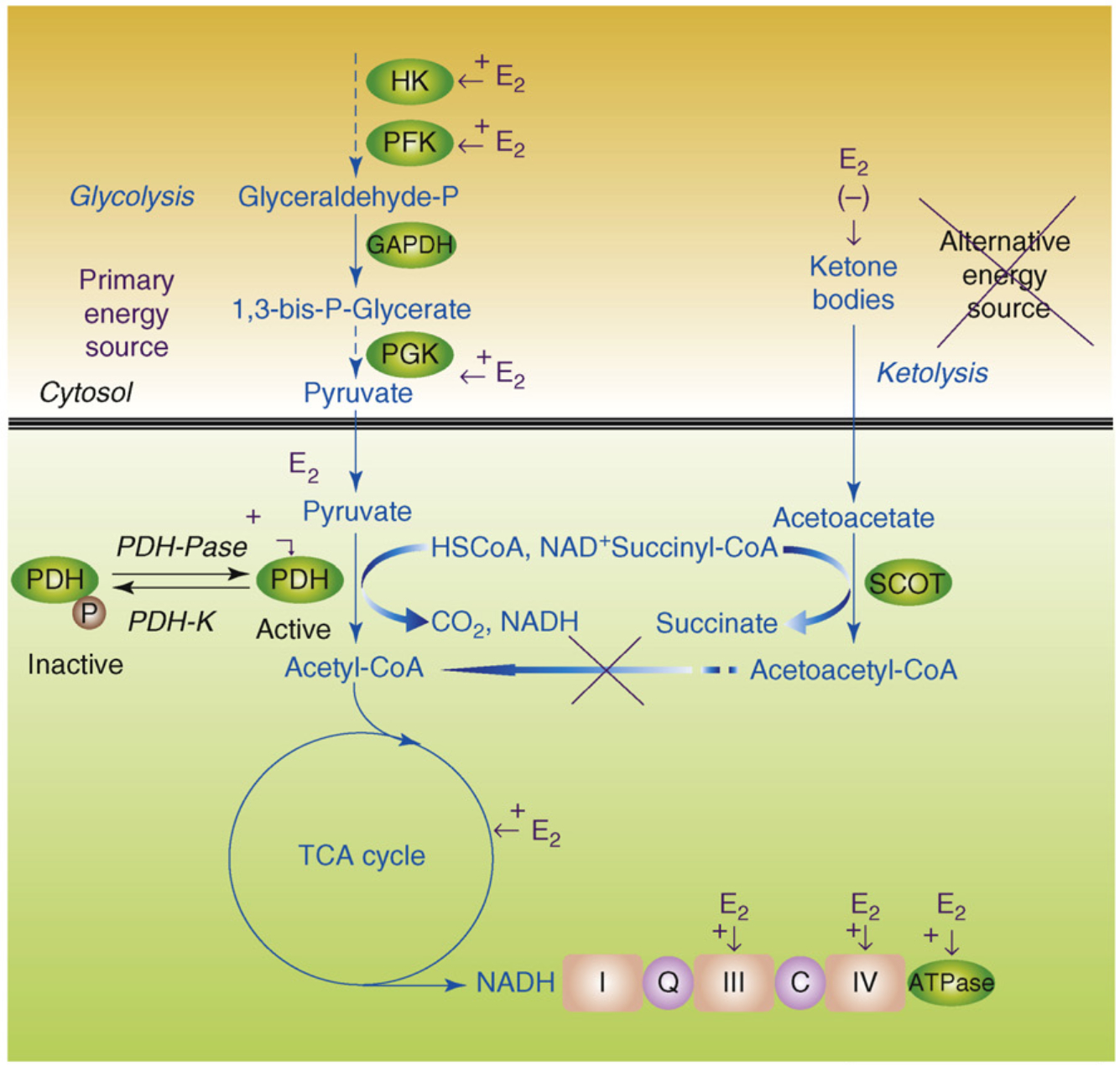
The 'healthy cell bias of estrogen action' hypothesis examines the role that regulating mitochondrial function and bioenergetics play in promoting neural health and the mechanistic crossroads that lead to divergent outcomes following estrogen exposure. Estrogen-induced signaling pathways in hippocampal and cortical neurons converge upon the mitochondria to enhance aerobic glycolysis coupled to the citric acid cycle, mitochondrial respiration and ATP generation. Convergence of estrogen-induced signaling onto mitochondria is also a point of vulnerability when activated in diseased neurons which exacerbates degeneration through increased load on dysregulated calcium homeostasis. As the continuum of neurological health progresses from healthy to unhealthy so too do the benefits of estrogen or hormone therapy. The healthy cell bias of estrogen action hypothesis provides a lens through which to assess disparities in outcomes across basic and clinical science and on which to predict outcomes of estrogen interventions for sustaining neurological health and preventing age-associated neurodegenerative diseases such as Alzheimer's.
Figures
References
-
- Brinton RD (2005) Investigative models for determining hormone therapy-induced outcomes in brain: evidence in support of a healthy cell bias of estrogen action. Ann. N. Y. Acad. Sci 1052, 57–74 - PubMed
-
- Wise PM (2006) Estrogen therapy: does it help or hurt the adult and aging brain? Insights derived from animal models. Neuroscience 138, 831–835 - PubMed
-
- Nilsen J and Brinton RD (2002) Impact of progestins on estrogen-induced neuroprotection: synergy by progesterone and 19-norprogesterone and antagonism by medroxyprogesterone acetate. Endocrinology 143, 205–212 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
