

Activation of peroxisome proliferator-activated receptor beta/delta induces lung cancer growth via peroxisome proliferator-activated receptor coactivator gamma-1alpha
- PMID: 18776129
- PMCID: PMC2645530
- DOI: 10.1165/rcmb.2008-0197OC
Activation of peroxisome proliferator-activated receptor beta/delta induces lung cancer growth via peroxisome proliferator-activated receptor coactivator gamma-1alpha
Retraction in
-
Retraction of two articles.Am J Respir Cell Mol Biol. 2012 Mar;46(3):414. doi: 10.1165/ajrcmb.46.3.414a. Am J Respir Cell Mol Biol. 2012. PMID: 22383655 Free PMC article. No abstract available.
Abstract
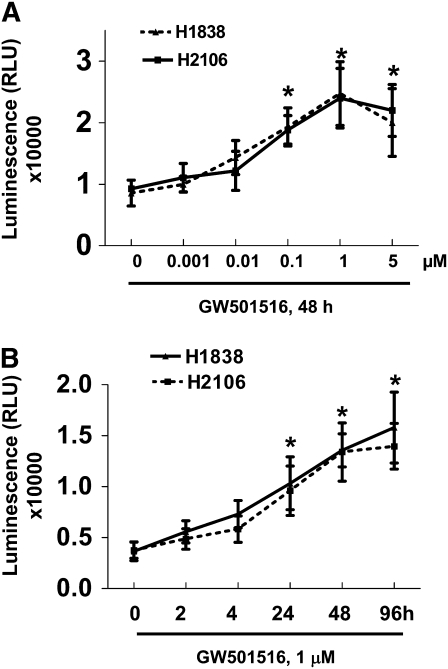
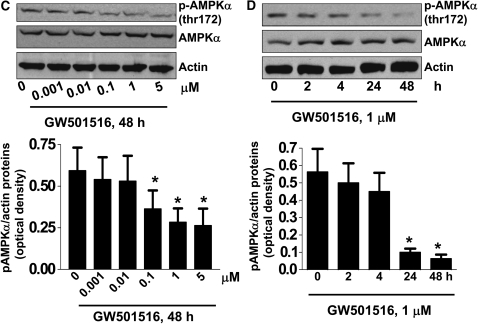
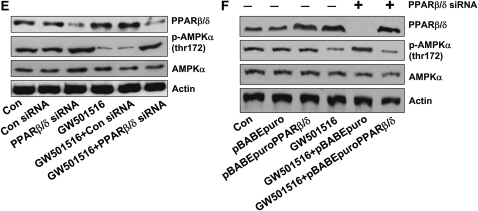
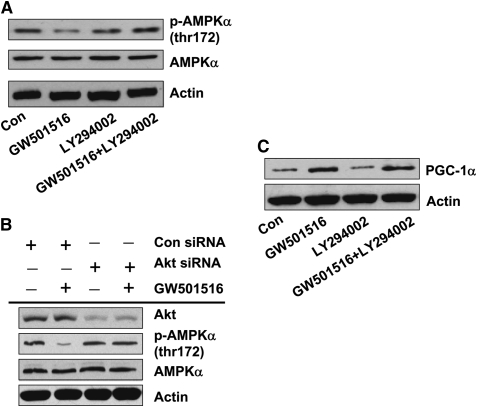
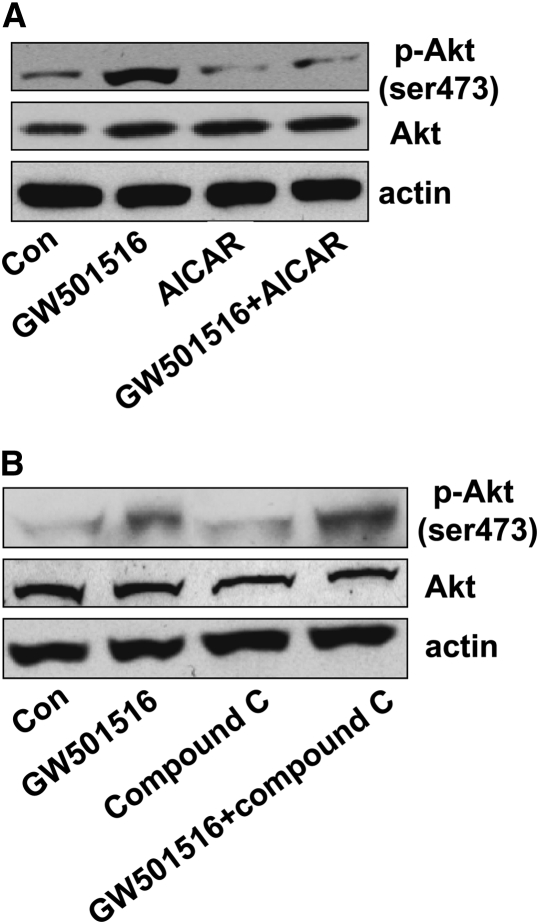
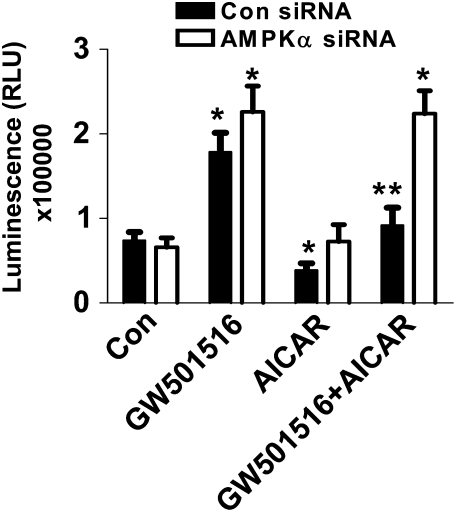
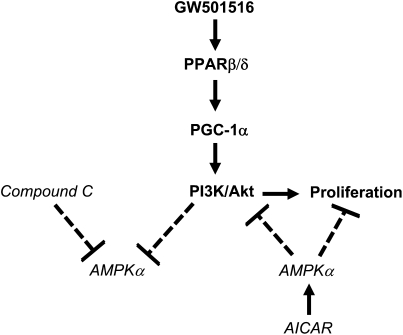
We previously demonstrated that a selective agonist of peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta), GW501516, stimulated human non-small cell lung carcinoma (NSCLC) growth, partly through inhibition of phosphatase and tensin homolog deleted on chromosome 10 expression. Here, we show that GW501516 also decreases the phosphorylation of AMP-activated protein kinase alpha (AMPKalpha), a major regulator of energy metabolism. This was mediated through specific activation of PPARbeta/delta, as a PPARbeta/delta small interfering RNA inhibited the effect. However, AMPKalpha did not mediate the growth-promoting effects of GW501516, as silencing of AMPKalpha did not inhibit GW501516-induced cell proliferation. Instead, we found that GW501516 stimulated peroxisome proliferator-activated receptor coactivator gamma (PGC)-1alpha, which activated the phosphatidylinositol 3 kinase (PI3-K)/Akt mitogenic pathway. An inhibitor of PI3-K, LY294002, had no effect on PGC-1alpha, consistent with PGC-1alpha being upstream of PI3-K/Akt. Of note, an activator of AMPKalpha, 5-amino-4-imidazole carboxamide riboside, inhibited the growth-promoting effects of GW501516, suggesting that although AMPKalpha is not responsible for the mitogenic effects of GW501516, its activation can oppose these events. This study unveils a novel mechanism by which GW501516 and activation of PPARbeta/delta stimulate human lung carcinoma cell proliferation, and suggests that activation of AMPKalpha may oppose this effect.
Figures

References
-
- Felip E, Rosell R. Testing for excision repair cross-complementing 1 in patients with non–small-cell lung cancer for chemotherapy response. Expert Rev Mol Diagn 2007;7:261–268. - PubMed
-
- Granville CA, Dennis PA. An overview of lung cancer genomics and proteomics. Am J Respir Cell Mol Biol 2005;32:169–176. - PubMed
-
- Petrashevskaya NN, Schwarz A. Peroxisome proliferator-activated receptor β/δ: a new antihypertrophic drug target? Cardiovasc Res 2005;65:770–771. - PubMed
-
- Braissant O, Wahli W. Differential expression of peroxisome proliferator-activated receptor-α, -β, and -γ during rat embryonic development. Endocrinology 1998;139:2748–2754. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
