

Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes
- PMID: 18776137
- PMCID: PMC2584123
- DOI: 10.2337/db08-0645
Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes
Abstract
Objective: The complexity of interactions between genes and the environment is a major challenge for type 1 diabetes studies. Nuclear chromatin is the interface between genetics and environment and the principal carrier of epigenetic information. Because histone tail modifications in chromatin are linked to gene transcription, we hypothesized that histone methylation patterns in cells from type 1 diabetic patients can provide novel epigenetic insights into type 1 diabetes and its complications.
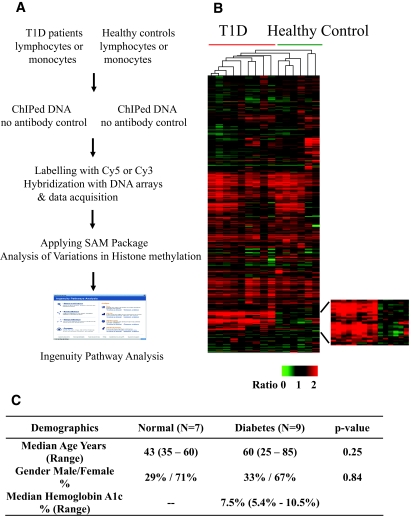
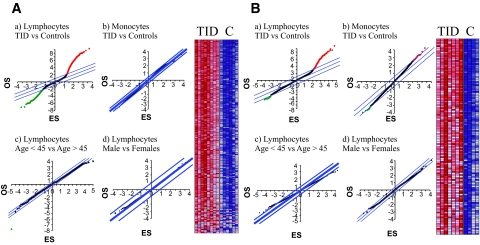
Research design and methods: We used chromatin immunoprecipitation (ChIP) linked to microarray (ChIP-chip) approach to compare genome-wide histone H3 lysine 9 dimethylation (H3K9me2) patterns in blood lymphocytes and monocytes from type 1 diabetic patients versus healthy control subjects. Bioinformatics evaluation of methylated candidates was performed by Ingenuity Pathway Analysis (IPA) tools.
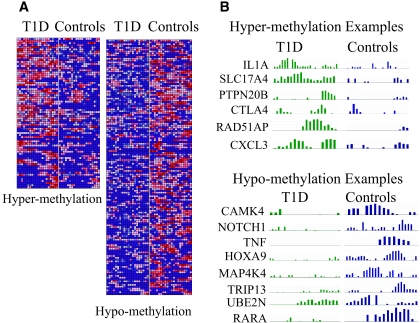
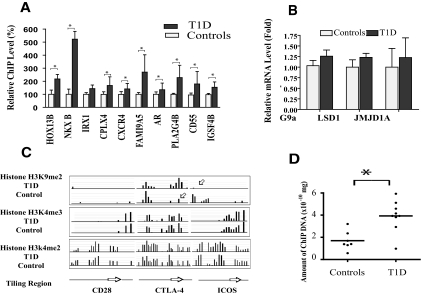
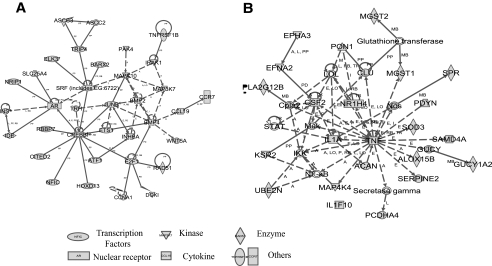
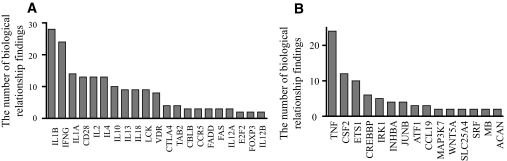
Results: A subset of genes in the type 1 diabetic cohort showed significant increase in H3K9me2 in lymphocytes but not in monocytes. CLTA4, a type 1 diabetes susceptibility gene, was one of the candidates displaying increased promoter H3K9me2 in type 1 diabetes. IPA identified two high-scoring networks that encompassed genes showing altered H3K9me2. Many of them were associated with autoimmune and inflammation-related pathways, such as transforming growth factor-beta, nuclear factor-kappaB, p38 mitogen-activated protein kinase, toll-like receptor, and interleukin-6. IPA also revealed biological relationships between these networks and known type 1 diabetes candidate genes.
Conclusions: The concerted and synergistic alteration of histone methylation within the identified network in lymphocytes might have an effect on the etiology of type 1 diabetes and its complications. These studies provide evidence of a novel association between type 1 diabetes and altered histone methylation of key genes that are components of type 1 diabetes-related biological pathways and also a new understanding of the pathology of type 1 diabetes.
Figures
Comment in
-
Immunopathogenic interaction of environmental triggers and genetic susceptibility in diabetes: is epigenetics the missing link?Diabetes. 2008 Dec;57(12):3184-6. doi: 10.2337/db08-1275. Diabetes. 2008. PMID: 19033405 Free PMC article. No abstract available.
References
-
- Davies JL, Kawaguchi Y, Bennett ST, Copeman JB, Cordell HJ, Pritchard LE, Reed PW, Gough SC, Jenkins SC, Palmer SM, Balfour KM, Rowe BR, Farrall M, Barnett AH, Bain SC, Todd JA: A genome-wide search for human type 1 diabetes susceptibility genes. Nature 371 :130 –136,1994 - PubMed
-
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, Lowe CE, Szeszko JS, Hafler JP, Zeitels L, Yang JH, Vella A, Nutland S, Stevens HE, Schuilenburg H, Coleman G, Maisuria M, Meadows W, Smink LJ, Healy B, Burren OS, Lam AA, Ovington NR, Allen J, Adlem E, Leung HT, Wallace C, Howson JM, Guja C, Ionescu-Tirgoviste C, Simmonds MJ, Heward JM, Gough SC, Dunger DB, Wicker LS, Clayton DG: Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet 39 :857 –864,2007 - PMC - PubMed
-
- An Essay by Dr. John Todd [article online],2002. . Available from http://www.esi-topics.com/diabetes/interviews/DrJohnTodd.html
-
- Klug A, Rhodes D, Smith J, Finch JT, Thomas JO: A low resolution structure for the histone core of the nucleosome. Nature 287 :509 –516,1980 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
