

Beta-cell failure as a complication of diabetes
- PMID: 18777097
- PMCID: PMC4456188
- DOI: 10.1007/s11154-008-9101-5
Beta-cell failure as a complication of diabetes
Abstract
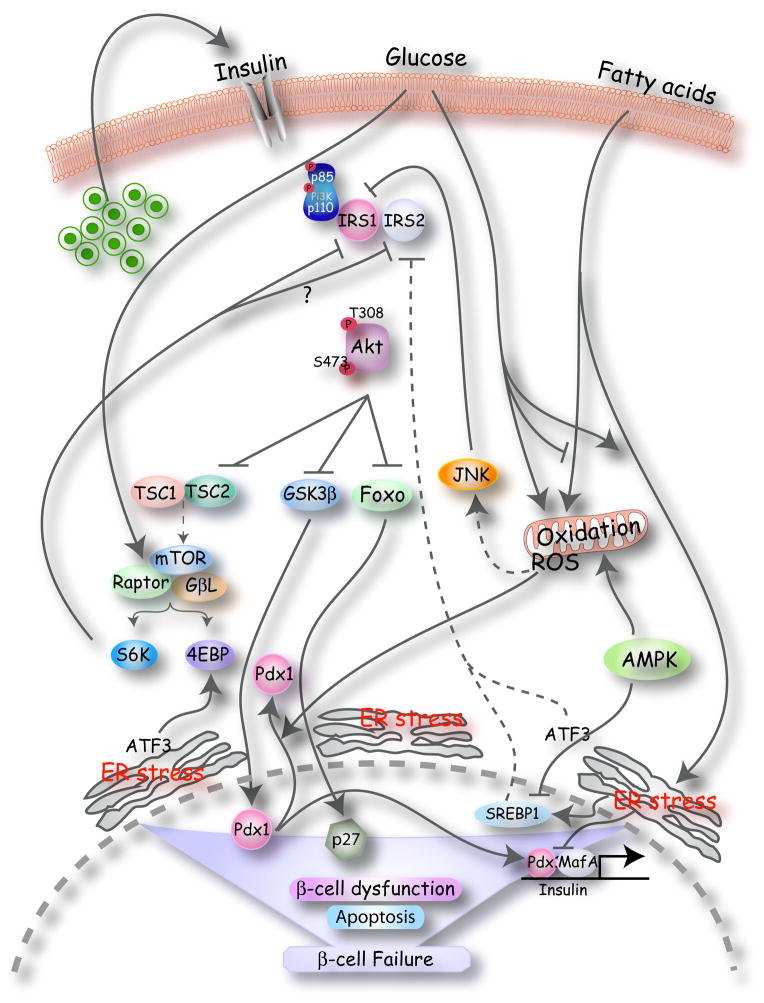
Type 2 diabetes mellitus is a complex disease characterized by beta-cell failure in the setting of insulin resistance. In early stages of the disease, pancreatic beta-cells adapt to insulin resistance by increasing mass and function. As nutrient excess persists, hyperglycemia and elevated free fatty acids negatively impact beta-cell function. This happens by numerous mechanisms, including the generation of reactive oxygen species, alterations in metabolic pathways, increases in intracellular calcium and the activation of endoplasmic reticulum stress. These processes adversely affect beta-cells by impairing insulin secretion, decreasing insulin gene expression and ultimately causing apoptosis. In this review, we will first discuss the regulation of beta-cell mass during normal conditions. Then, we will discuss the mechanisms of beta-cell failure, including glucotoxicity, lipotoxicity and endoplasmic reticulum stress. Further research into mechanisms will reveal the key modulators of beta-cell failure and thus identify possible novel therapeutic targets. Type 2 diabetes mellitus is a multifactorial disease that has greatly risen in prevalence in part due to the obesity and inactivity that characterize the modern Western lifestyle. Pancreatic beta-cells possess the potential to greatly expand their function and mass in both physiologic and pathologic states of nutrient excess and increased insulin demand. beta-cell response to nutrient excess occurs by several mechanisms, including hypertrophy and proliferation of existing beta-cells, increased insulin production and secretion, and formation of new beta-cells from progenitor cells [1, 2]. Failure of pancreatic beta-cells to adequately expand in settings of increased insulin demand results in hyperglycemia and diabetes. In this review, we will first discuss the factors involved in beta-cell growth and then discuss the mechanisms by which beta-cell expansion fails and leads to beta-cell failure and diabetes (Fig. 1).
Figures
References
-
- Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic beta-cells in adult rats after short-term glucose infusion. Diabetes. 1989;38:49–53. - PubMed
-
- Bruning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell. 1997;88:561–572. - PubMed
-
- Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology. 1992;130:1459–1466. - PubMed
-
- Scaglia L, Smith FE, Bonner-Weir S. Apoptosis contributes to the involution of beta cell mass in the post partum rat pancreas. Endocrinology. 1995;136:5461–5468. - PubMed
-
- Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren J-M, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, Bonner-Weir S, White MF. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–903. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
