

Analysis of polarization in QM/MM modelling of biologically relevant hydrogen bonds
- PMID: 18782723
- PMCID: PMC2585273
- DOI: 10.1098/rsif.2008.0243.focus
Analysis of polarization in QM/MM modelling of biologically relevant hydrogen bonds
Abstract
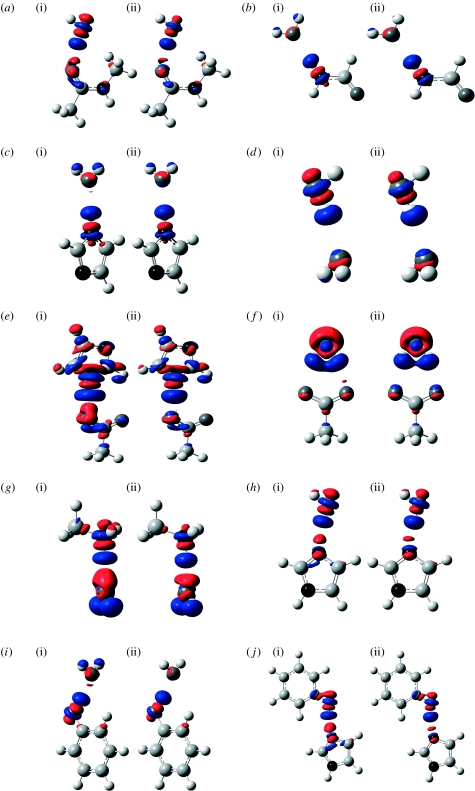
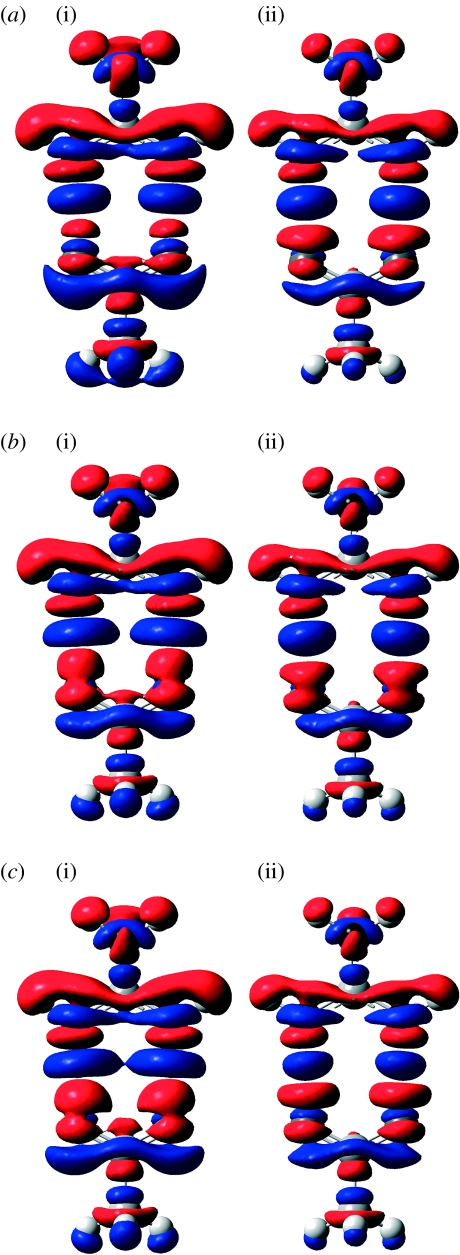
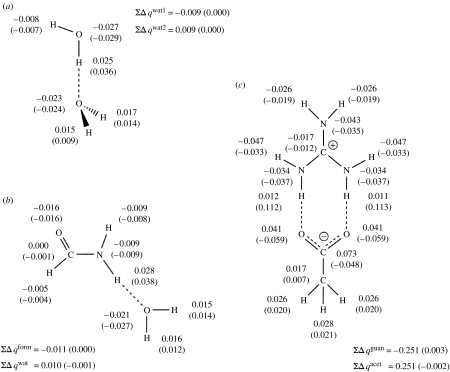
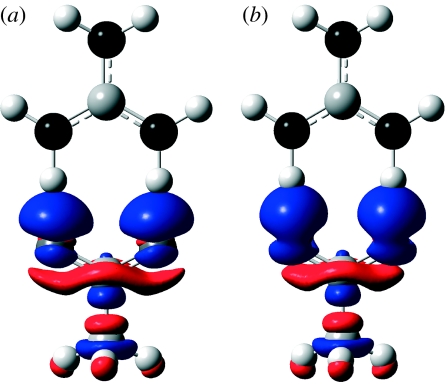
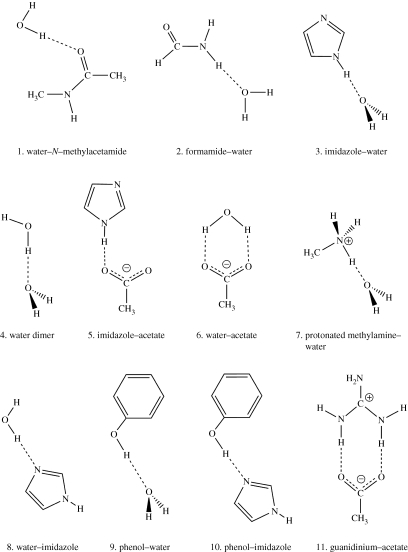
Combined quantum mechanics/molecular mechanics (QM/MM) methods are increasingly important for the study of chemical reactions and systems in condensed phases. Here, we have tested the accuracy of a density functional theory-based QM/MM implementation (B3LYP/6-311+G(d,p)/CHARMM27) on a set of biologically relevant interactions by comparison with full QM calculations. Intermolecular charge transfer due to hydrogen bond formation is studied to assess the severity of spurious polarization of QM atoms by MM point charges close to the QM/MM boundary. The changes in total electron density and natural bond orbital atomic charges due to hydrogen bond formation in selected complexes obtained at the QM/MM level are compared with full QM results. It is found that charge leakage from the QM atoms to MM atomic point charges close to the QM/MM boundary is not a serious problem, at least with limited basis sets. The results are encouraging in showing that important properties of key biomolecular interactions can be treated well at the QM/MM level employing good-quality levels of QM theory.
Figures
Similar articles
-
Redistributed charge and dipole schemes for combined quantum mechanical and molecular mechanical calculations.J Phys Chem A. 2005 May 5;109(17):3991-4004. doi: 10.1021/jp0446332. J Phys Chem A. 2005. PMID: 16833721
-
Comparison of polarizable continuum model and quantum mechanics/molecular mechanics solute electronic polarization: study of the optical and magnetic properties of diazines in water.J Chem Phys. 2011 Oct 14;135(14):144103. doi: 10.1063/1.3644894. J Chem Phys. 2011. PMID: 22010694
-
Lennard-Jones Parameters for B3LYP/CHARMM27 QM/MM Modeling of Nucleic Acid Bases.J Chem Theory Comput. 2009 Feb 10;5(2):396-410. doi: 10.1021/ct800135k. J Chem Theory Comput. 2009. PMID: 26610113
-
Biomolecular force fields: where have we been, where are we now, where do we need to go and how do we get there?J Comput Aided Mol Des. 2019 Feb;33(2):133-203. doi: 10.1007/s10822-018-0111-4. Epub 2018 Nov 30. J Comput Aided Mol Des. 2019. PMID: 30506158 Review.
-
QM/MM Calculations on Proteins.Methods Enzymol. 2016;577:119-58. doi: 10.1016/bs.mie.2016.05.014. Epub 2016 Jun 28. Methods Enzymol. 2016. PMID: 27498637 Review.
Cited by
-
Does compound I vary significantly between isoforms of cytochrome P450?J Am Chem Soc. 2011 Oct 5;133(39):15464-74. doi: 10.1021/ja203157u. Epub 2011 Sep 12. J Am Chem Soc. 2011. PMID: 21863858 Free PMC article.
-
Electronic Absorption Spectra from MM and ab initio QM/MM Molecular Dynamics: Environmental Effects on the Absorption Spectrum of Photoactive Yellow Protein.J Chem Theory Comput. 2012 Dec 1;8(12):5092-5106. doi: 10.1021/ct3006826. Epub 2012 Oct 6. J Chem Theory Comput. 2012. PMID: 23476156 Free PMC article.
-
Introduction. Biomolecular simulation.J R Soc Interface. 2008 Dec 6;5 Suppl 3(Suppl 3):S169-72. doi: 10.1098/rsif.2008.0385.focus. J R Soc Interface. 2008. PMID: 18826912 Free PMC article.
-
Nanotechnology-Driven Drug Delivery Systems for Lung Cancer: Computational Advances and Clinical Perspectives.Thorac Cancer. 2025 Jul;16(14):e70134. doi: 10.1111/1759-7714.70134. Thorac Cancer. 2025. PMID: 40682256 Free PMC article. Review.
-
Calculation of chromophore excited state energy shifts in response to molecular dynamics of pigment-protein complexes.Photosynth Res. 2011 Oct;110(1):25-38. doi: 10.1007/s11120-011-9689-2. Epub 2011 Oct 1. Photosynth Res. 2011. PMID: 21964859
References
-
- Antes I., Thiel W. Adjusted connection atoms for combined quantum mechanical and molecular mechanical methods. J. Phys. Chem. A. 1999;103:9290–9295. doi: 10.1021/jp991771w. - DOI
-
- Aqvist J., Warshel A. Simulation of enzyme-reactions using valence-bond force-fields and other hybrid quantum-classical approaches. Chem. Rev. 1993;93:2523–2544. doi: 10.1021/cr00023a010. - DOI
-
- Assfeld X., Rivail J.L. Quantum chemical computations on parts of large molecules: the ab-initio local self consistent field method. Chem. Phys. Lett. 1996;263:100–106. doi: 10.1016/S0009-2614(96)01165-7. - DOI
-
- Bakowies D., Thiel W. Hybrid models for combined quantum mechanical and molecular mechanical approaches. J. Phys. Chem. A. 1996;100:10 580–10 594. doi: 10.1021/jp9536514. - DOI
-
- Bartha F., Kapuy O., Kozmutza C., Van Alsenoy C. Analysis of weakly bound structures: hydrogen bond and the electron density in a water dimer. J. Mol. Struct. (Theochem.) 2003;666:117–122. doi: 10.1016/j.theochem.2003.08.020. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
