

Feature context-dependency and complexity-reduction in probability landscapes for integrative genomics
- PMID: 18783599
- PMCID: PMC2559821
- DOI: 10.1186/1742-4682-5-21
Feature context-dependency and complexity-reduction in probability landscapes for integrative genomics
Abstract
Background: The question of how to integrate heterogeneous sources of biological information into a coherent framework that allows the gene regulatory code in eukaryotes to be systematically investigated is one of the major challenges faced by systems biology. Probability landscapes, which include as reference set the probabilistic representation of the genomic sequence, have been proposed as a possible approach to the systematic discovery and analysis of correlations amongst initially heterogeneous and un-relatable descriptions and genome-wide measurements. Much of the available experimental sequence and genome activity information is de facto, but not necessarily obviously, context dependent. Furthermore, the context dependency of the relevant information is itself dependent on the biological question addressed. It is hence necessary to develop a systematic way of discovering the context-dependency of functional genomics information in a flexible, question-dependent manner.
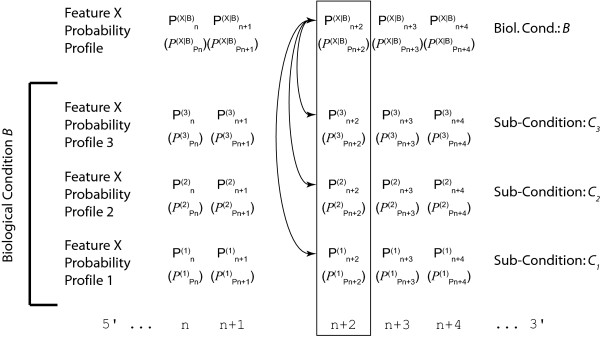
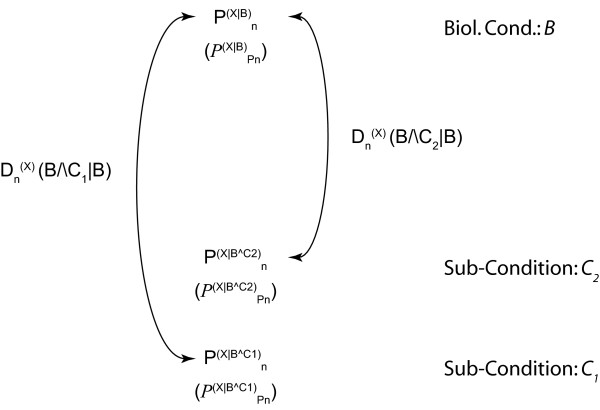
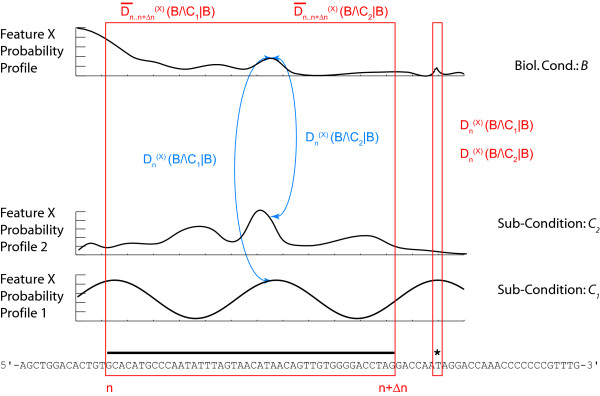
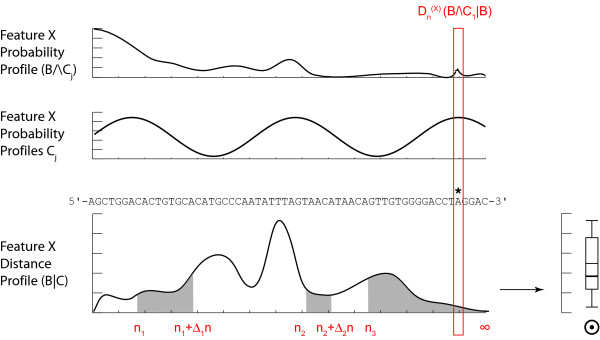
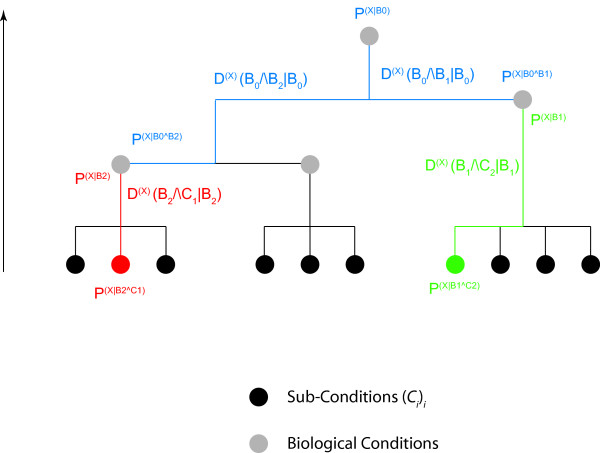
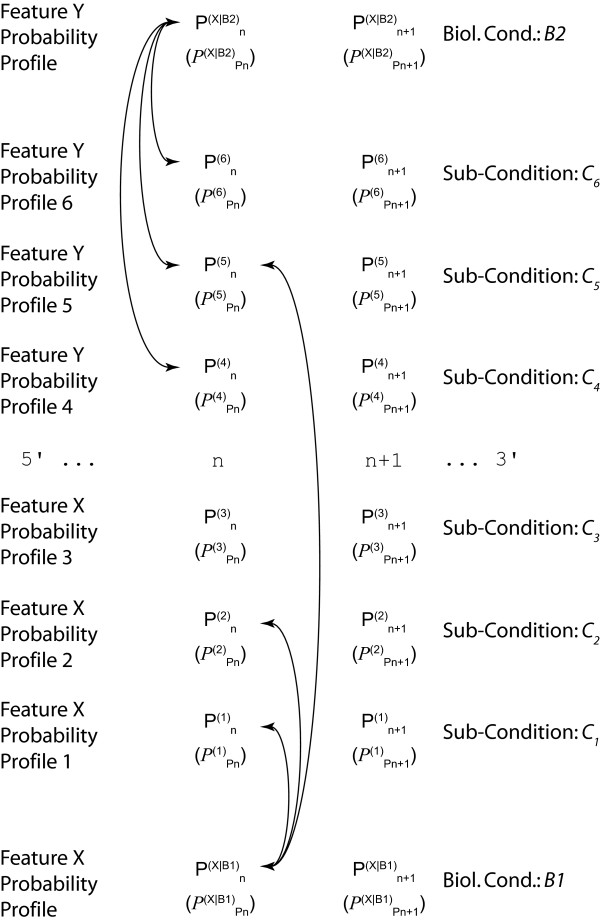
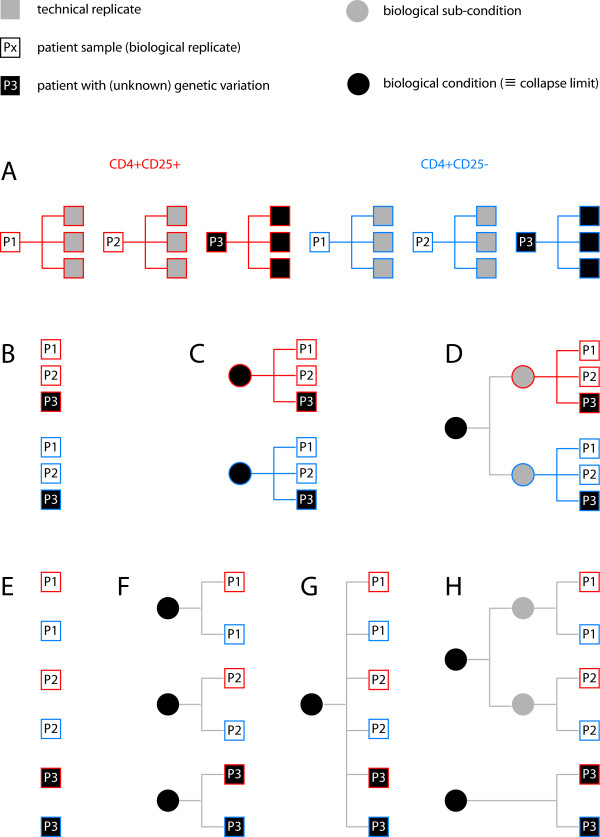
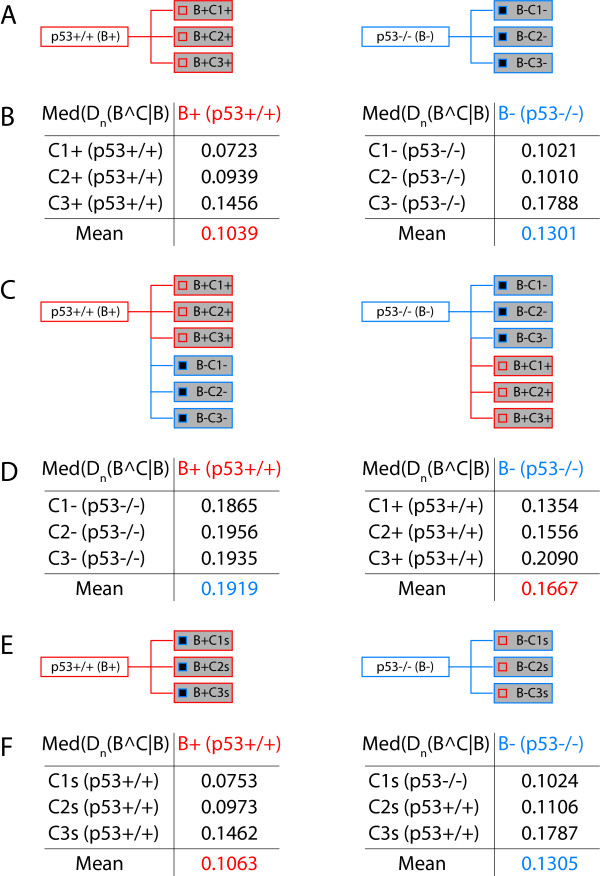
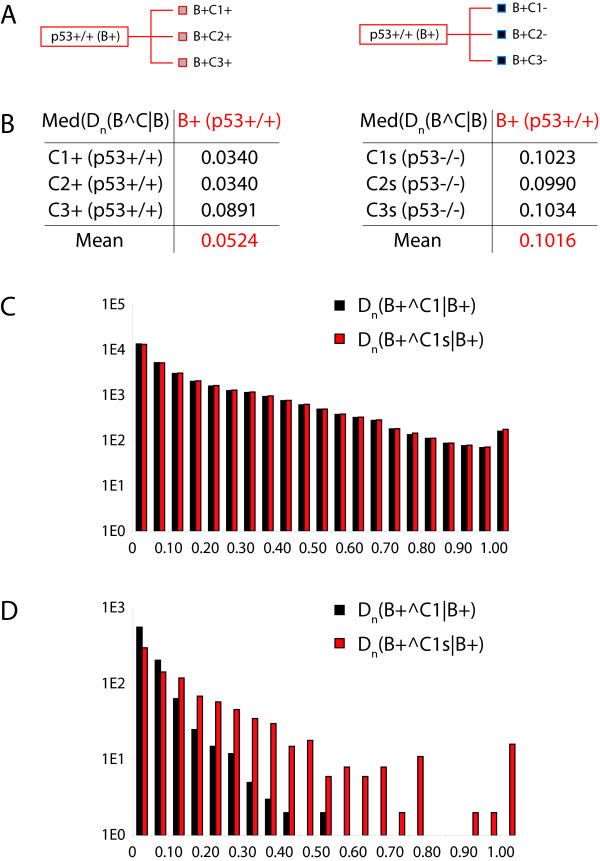
Results: We demonstrate here how feature context-dependency can be systematically investigated using probability landscapes. Furthermore, we show how different feature probability profiles can be conditionally collapsed to reduce the computational and formal, mathematical complexity of probability landscapes. Interestingly, the possibility of complexity reduction can be linked directly to the analysis of context-dependency.
Conclusion: These two advances in our understanding of the properties of probability landscapes not only simplify subsequent cross-correlation analysis in hypothesis-driven model building and testing, but also provide additional insights into the biological gene regulatory problems studied. Furthermore, insights into the nature of individual features and a classification of features according to their minimal context-dependency are achieved. The formal structure proposed contributes to a concrete and tangible basis for attempting to formulate novel mathematical structures for describing gene regulation in eukaryotes on a genome-wide scale.
Figures
Similar articles
-
Probability landscapes for integrative genomics.Theor Biol Med Model. 2008 May 20;5:9. doi: 10.1186/1742-4682-5-9. Theor Biol Med Model. 2008. PMID: 18492240 Free PMC article.
-
Describing the complexity of systems: multivariable "set complexity" and the information basis of systems biology.J Comput Biol. 2014 Feb;21(2):118-40. doi: 10.1089/cmb.2013.0039. Epub 2013 Dec 30. J Comput Biol. 2014. PMID: 24377753 Free PMC article.
-
Integration of multi-omics data of a genome-reduced bacterium: Prevalence of post-transcriptional regulation and its correlation with protein abundances.Nucleic Acids Res. 2016 Feb 18;44(3):1192-202. doi: 10.1093/nar/gkw004. Epub 2016 Jan 14. Nucleic Acids Res. 2016. PMID: 26773059 Free PMC article.
-
Functional genomics and proteomics in the clinical neurosciences: data mining and bioinformatics.Prog Brain Res. 2006;158:83-108. doi: 10.1016/S0079-6123(06)58004-5. Prog Brain Res. 2006. PMID: 17027692 Review.
-
Neuroscience in the era of functional genomics and systems biology.Nature. 2009 Oct 15;461(7266):908-15. doi: 10.1038/nature08537. Nature. 2009. PMID: 19829370 Free PMC article. Review.
Cited by
-
The R2R3-MYB, bHLH, WD40, and related transcription factors in flavonoid biosynthesis.Funct Integr Genomics. 2013 Mar;13(1):75-98. doi: 10.1007/s10142-012-0301-4. Epub 2012 Nov 27. Funct Integr Genomics. 2013. PMID: 23184474
-
Critical dynamics in host-pathogen systems.Curr Top Microbiol Immunol. 2013;363:235-59. doi: 10.1007/82_2012_260. Curr Top Microbiol Immunol. 2013. PMID: 22976347 Free PMC article. Review.
-
Dynamics of DNA damage induced pathways to cancer.PLoS One. 2013 Sep 4;8(9):e72303. doi: 10.1371/journal.pone.0072303. eCollection 2013. PLoS One. 2013. PMID: 24023735 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
