

doi: 10.1021/jm800562d.
Epub 2008 Sep 12.
Target flexibility: an emerging consideration in drug discovery and design
Affiliations
- PMID: 18785728
- PMCID: PMC2701403
- DOI: 10.1021/jm800562d
Item in Clipboard
Target flexibility: an emerging consideration in drug discovery and design
J Med Chem.
.
No abstract available
Figures
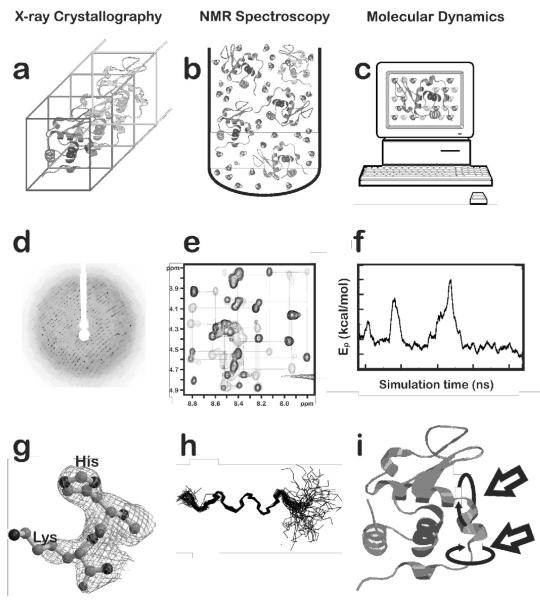
X-ray crystallography, NMR spectroscopy and computational Molecular Dynamics. a) A crystal illustrating the uniformity of the protein molecular structures within their unit cells necessary for X-ray diffraction. Ordered water molecules (not shown) partially hydrate the structure; b) protein molecule in solution for NMR experiment. Ordered water and other (not shown) ions solvate the structure, but many other solvent molecules are not structurally ordered; c) protein immersed in virtual (water) solvent for computational Molecular Dynamics with periodic boundary conditions; d) diffraction pattern from an X-ray data collection; e) typical 2D NOESY spectrum (http://www.sanger.ac.uk/Users/sgj/thesis/html/node86.html >) from protein NMR; f) Molecular Dynamics potential energy as a function of simulation time for coarse-grained motions; g) electron density maps for histidine and lysine residues. The density around more labile (high B-factor) atoms is either diffuse or non-existent (e.g., the NZ atom of lysine); h) typical backbone traces for structures meeting constraints determined by NMR NOESY experiments. The high degree of flexibility at either end of the structure is evident; and i) protein structure illustrating (see arrows) typical large scale motions that may be observed with computational Molecular Dynamics.
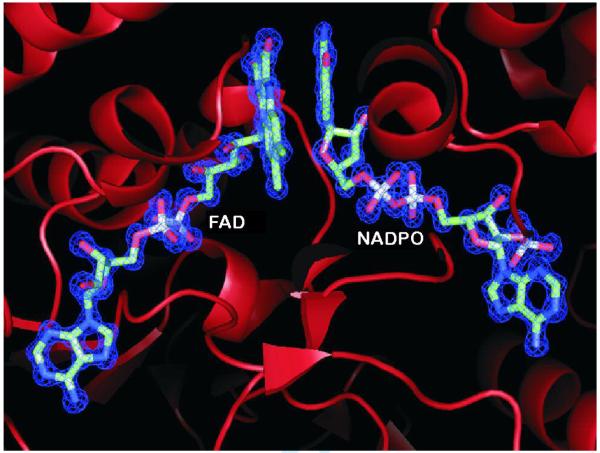
View of the active site of Mycobacterium tuberculosis FprA. The FAD cofactor and a covalently modified NADP+ (labeled NADPO) identified in the 1.05 Å resolution crystal structure (PDB code, 1LQT) are highlighted. Shown in blue is the 2Fo-Fc map contoured at the level of 2 σ above its mean. (Prepared using PyMOL.)
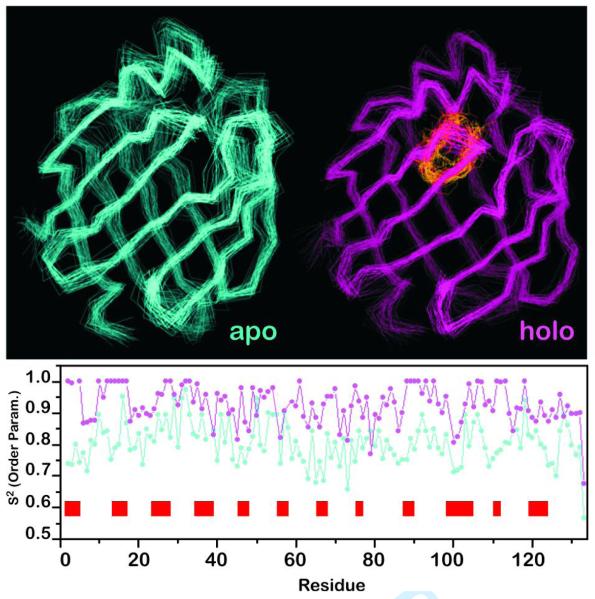
Backbone dynamics data derived from 15N-1H NMR experiments on mutant Sm14-M20(C62V). The NMR-derived backbone traces for both apo and holo protein forms are shown on top, with the bound fatty acid shown in yellow (in the holo form). The order parameters S2 as functions of residue number are shown in the bottom graph for apo (cyan) and holo (magenta). The red bars along the residue axis indicate the loop regions of the protein, where a larger difference in S2 between the two forms is observed.
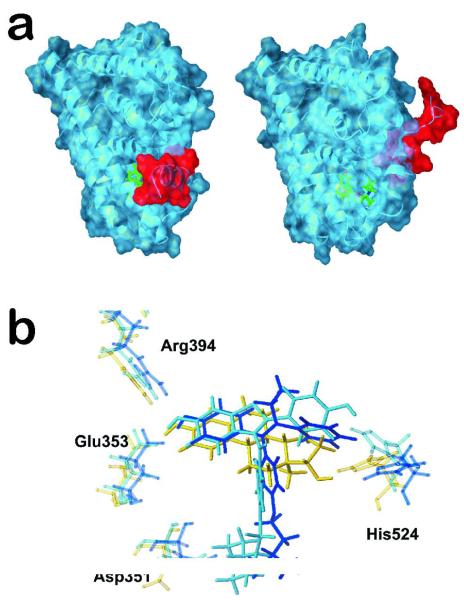
Examples of large motions (coarse-grained) and small motions in the estrogen receptor α protein. a) The different positions of helix 12 (red) depending of the type of bound ligand (green): agonists such as estradiol induce and stabilize the closed conformation (left) while antagonists such as tamoxifen prevent helix 12 from adopting the agonist-induced conformation (right); b) small adjustments of the His524 residue within the ERα binding site depending on the ligand. Yellow bonds indicate the positions of His524 when the natural ligand estradiol (also yellow) is bound; light blue bonds illustrate the antitumor drug raloxifen and its effect on His524; dark blue bonds represents the drug tamoxifen and its effect on His524.
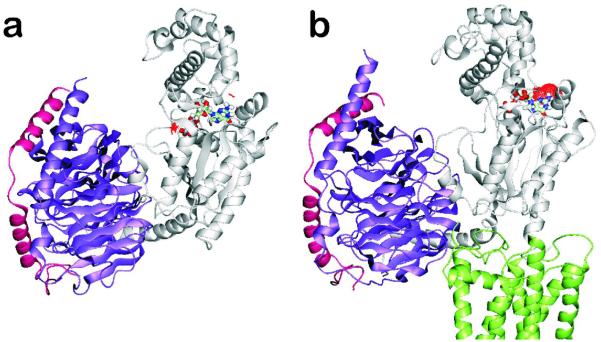
Average minimized structures of a) free and b) TXA2-bound heterotrimeric Gq. TXA2 and the G protein α-, β- and γ-subunits are, respectively, colored in green, gray, violet and magenta. The GDP molecule is colored by atom type. Red dots indicate the solvent accessible surface of GDP, which is exposed to solvent upon receptor binding. Only the intracellular half of the receptor is shown.
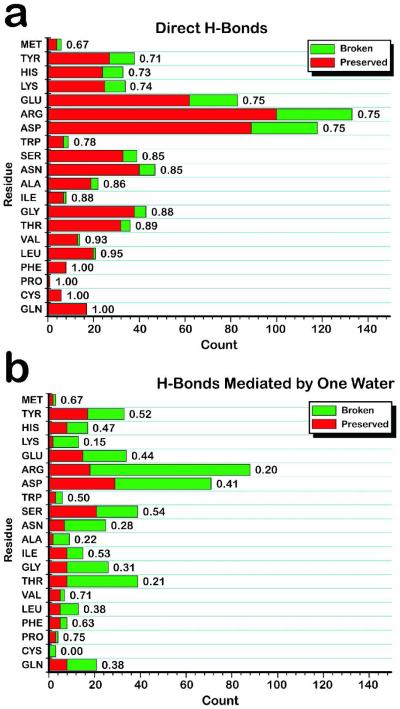
a) Direct intra-protein hydrogen bonds tend to be preserved upon ligand binding. Ligand-free and ligand-bound crystal structures for 32 proteins with low pairwise sequence identity (<30%) were analyzed. The percentage of direct, intra-protein hydrogen bonds preserved upon ligand binding (red) is compared with the percentage broken (green) for each residue type, including main-chain and side-chain hydrogen bonds. Typically 75% or more of direct hydrogen bonds are preserved. b) Intra-protein water-mediated hydrogen bonds tend to break upon ligand binding. Analysis was performed for intra-protein hydrogen bonds mediated by one water molecule. The trend is opposite to that found for direct hydrogen bonds: 50-80% of water-mediated hydrogen bonds are broken upon ligand binding. Details: All residues containing an atom ≤4 Å from the ligand (in the ligand-bound structure, or ≤ 4 Å from the ligand superimposed into the ligand-free structure) or within 4 Å of a water molecule bridging between the protein and ligand, were kept for analysis. Intra-protein hydrogen bonds were initially identified as having a donor-acceptor distance ≤ 3.6 Å, hydrogen-acceptor distance ≤ 2.6 Å, and donor-H-acceptor angle of 90-180°. This set was screened by a hydrogen bond energy function evaluating detailed atom chemistry-dependent features, to ensure that very weak hydrogen bonds were excluded. 60 of the 64 structures had resolution of 2.2 Å or better, and the remaining 4 had resolution ≤ 2.6 Å.
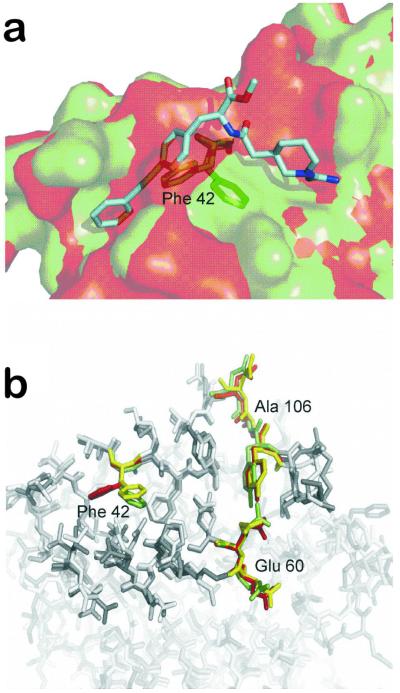
Adaptive nature of the protein-protein interface of the cytokine interleukin-2. a) Overlay of the unbound (red) (PDB code: 1M47) and bound (green) (PDB code: 1M48) protein structure of IL-2 together with a small molecule (cyan) that buries into a groove not seen in the free structure of IL-2. Residue Phe42, whose resultant movement after small molecule binding opens the groove, is depicted in stick representation. b) Overlay of the protein-protein interface region (opaque sticks) of IL-2 in unbound (red) and bound (green) form. In addition, a snapshot from a FRODA simulation started from the unbound state is shown (yellow), which demonstrates that the movement of Phe42 can even be observed in the absence of the ligand, leading to a transient pocket opening. Interestingly, regions for which no movement was observed by experiment (around Glu60 and Ala106) also remain immobile during the simulation. (Pfleger, Metz, Kopitz and Gohlke, unpublished results.)
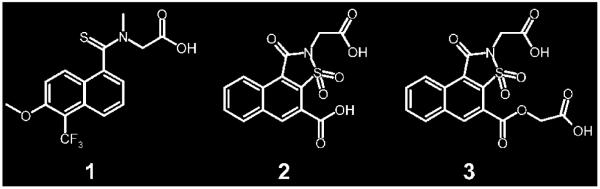
Significantly different binding-site conformations are induced in aldose reductase upon binding of the inhibitors tolrestat (1) and analogs (2,3).
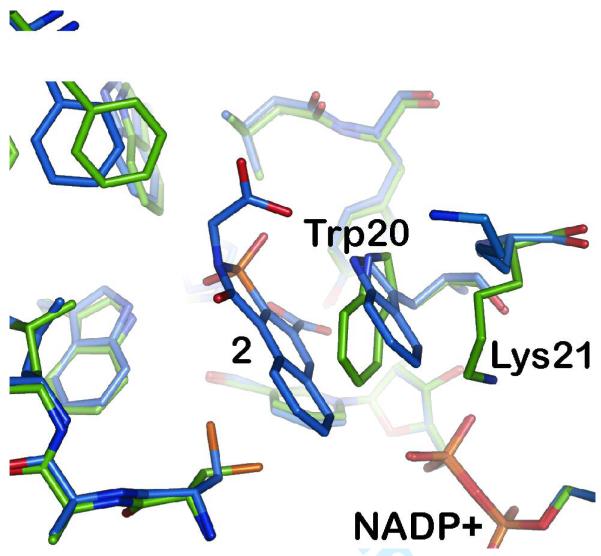
The complex of aldose reductase with 2 (blue) shows an unexpected conformational change in the binding site compared to the standard conformation previously observed (green). The side chain of Trp20 is rotated by 35° and, more importantly, the salt bridge between the side chain of Lys21 and the NADP+ cofactor is broken.
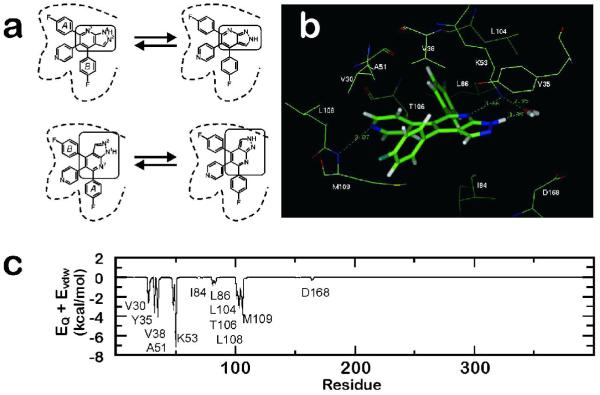
Determination of the binding mode of a pyridinyl-heterocycle inhibitor binding to p38 MAP kinase. a) The four possible binding modes of compound 4 corresponding to the two possible tautomers (N1-H (upper left) and N2-H (upper right)) and their respective pseudosymmetric arrangements arising from a 180° rotation around the pyridine-pyrazolopyridine axis (bottom). For clarity, the central core of the molecule is marked in all cases and key residues defining the binding pocket are displayed as reference; b) detail of the optimum binding mode for compound 4 derived from MD simulations; significant interactions, including a water bridge between 4 and Lys53, are shown; and c) interaction profile in kcal/mol for the sum of electrostatic and van der Waals energy for the residues of p38α MAP kinase and compound 4. Key residues for binding are noted. (Adapted from ref. .)
References
-
- Abraham DJ. Comprehensive Medicinal Chemistry II. Elsevier; Oxford: 2007. Structure-based Drug Design - A Historical Perspective and the Future.
-
- Bolton W, Perutz MF. Three dimensional fourier synthesis of horse deoxyhaemoglobin at 2.8 Angstrom units resolution. Nature. 1970;228:551–552. - PubMed
-
- Fermi G, Perutz MF, Shaanan B, Fourme R. The crystal structure of human deoxyhaemoglobin at 1.74 A resolution. J Mol Biol. 1984;175:159–174. - PubMed
-
- Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, et al. A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature. 1958;181:662–666. - PubMed
-
- Watson H. The Stereochemistry of the Protein Myoglobin. The Stereochemistry of the Protein Myoglobin. 1969;4:299–333.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
