

Target gene-specific regulation of androgen receptor activity by p42/p44 mitogen-activated protein kinase
- PMID: 18787043
- PMCID: PMC2582542
- DOI: 10.1210/me.2007-0481
Target gene-specific regulation of androgen receptor activity by p42/p44 mitogen-activated protein kinase
Abstract
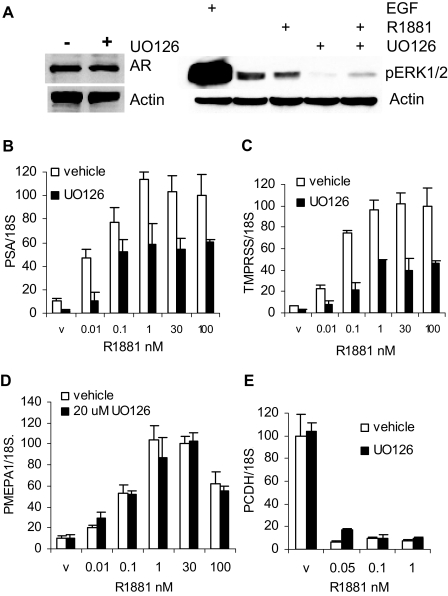
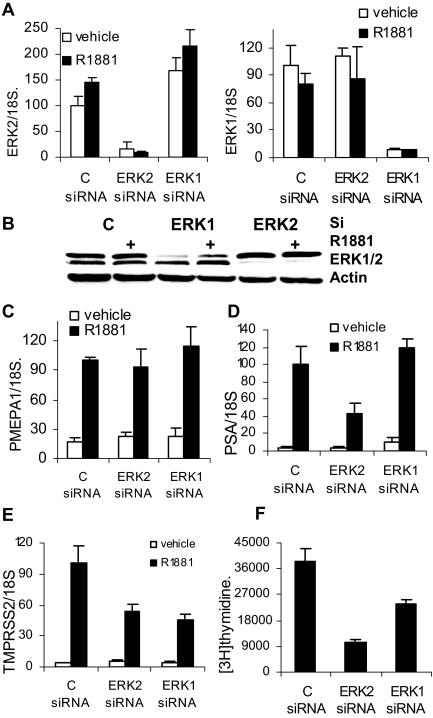
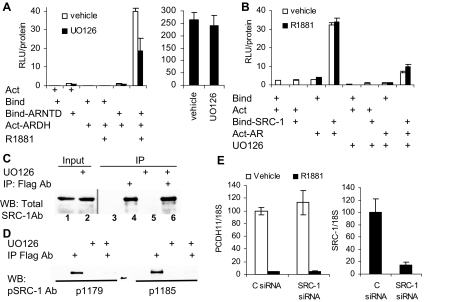
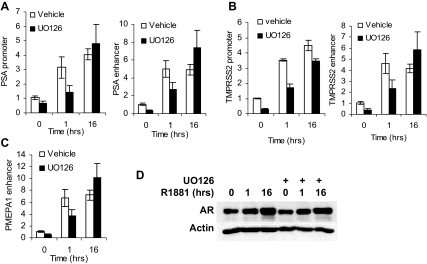
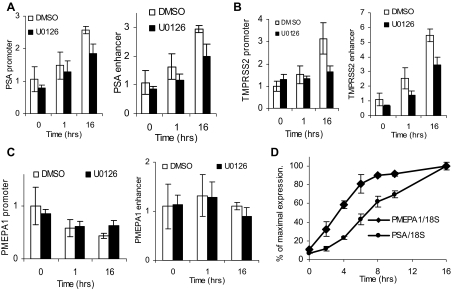
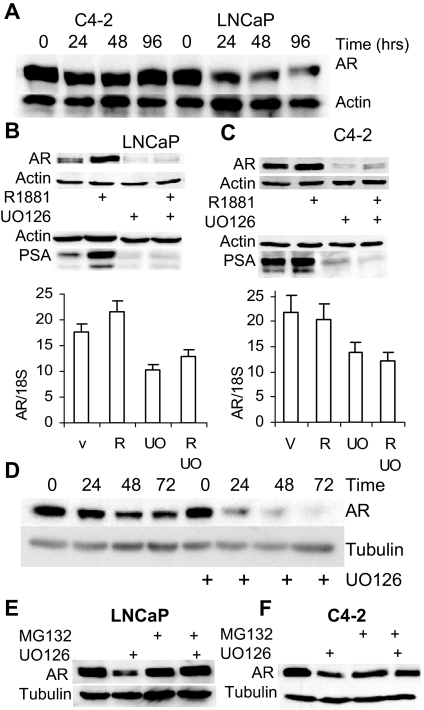
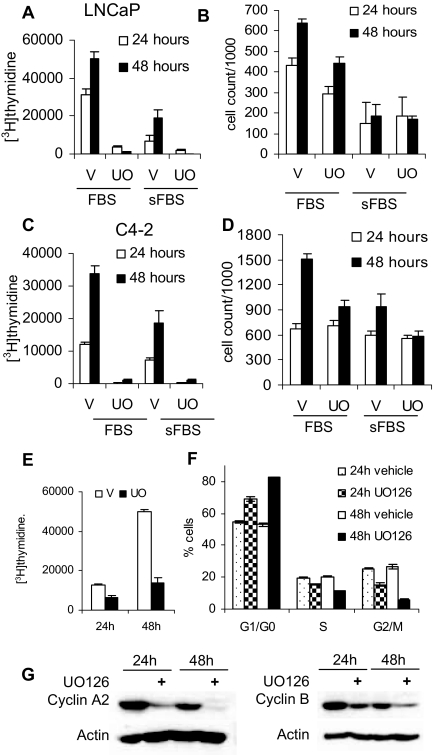
Evidence that the androgen receptor (AR) is not only important in androgen-dependent prostate cancer, but also continues to play a role in tumors that become resistant to androgen deprivation therapies, highlights the need to find alternate means to block AR activity. AR, a hormone-activated transcription factor, and its coactivators are phosphoproteins. Thus, we sought to determine whether inhibition of specific cell signaling pathways would reduce AR function. We found that short-term inhibition of p42/p44 MAPK activity either by a MAPK kinase inhibitor, U0126, or by depletion of kinase with small interfering RNA caused target gene-specific reductions in AR activity. AR enhances histone H3 acetylation of target genes that are sensitive to U0126 including prostate-specific antigen and TMPRSS2, but does not increase histone H3 acetylation of the U0126-resistant PMEPA1 gene. Thus, although AR induces transcription of many target genes, the molecular changes induced by AR at the chromatin level are target gene specific. Long-term treatment (24-48 h) with U0126 causes a G1 cell cycle arrest and reduces AR expression both through a decrease in AR mRNA and a reduction in AR protein stability. Thus, treatments that reduce p42/p44 MAPK activity in prostate cancer have the potential to reduce AR activity through a reduction in expression levels as well as by target gene-selective inhibition of AR function.
Figures
References
-
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM 2005 Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310:644–648 - PubMed
-
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL 2004 Molecular determinants of resistance to antiandrogen therapy. Nat Med 10:33–39 - PubMed
-
- Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ 2002 Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res 62:1008–1013 - PubMed
-
- Agoulnik IU, Vaid A, Bingman 3rd WE, Erdeme H, Frolov A, Smith CL, Ayala G, Ittmann MM, Weigel NL 2005 Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res 65:7959–7967 - PubMed
-
- Agoulnik IU, Weigel NL 2006 Androgen receptor action in hormone-dependent and recurrent prostate cancer. J Cell Biochem 99:362–372 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
