

Evolution of a new function by degenerative mutation in cephalochordate steroid receptors
- PMID: 18787702
- PMCID: PMC2527136
- DOI: 10.1371/journal.pgen.1000191
Evolution of a new function by degenerative mutation in cephalochordate steroid receptors
Abstract
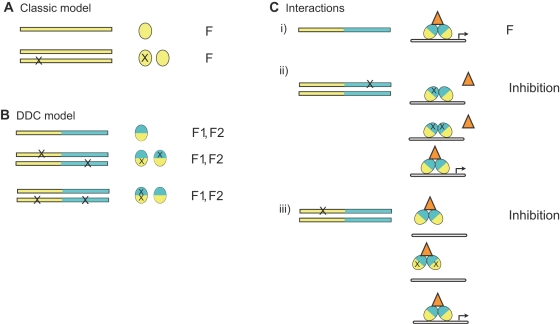
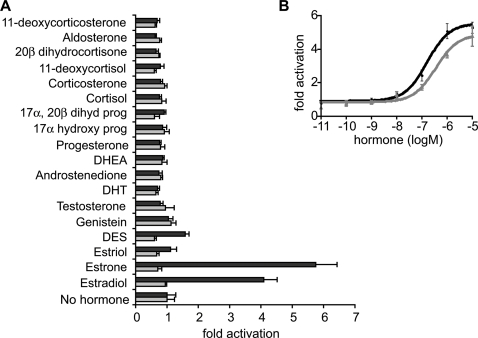
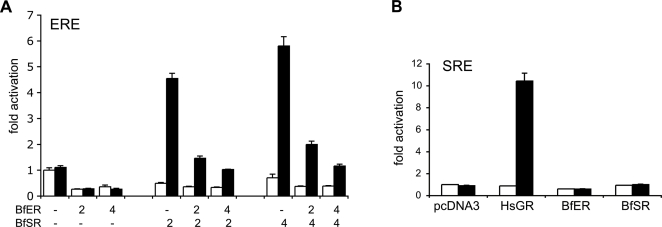
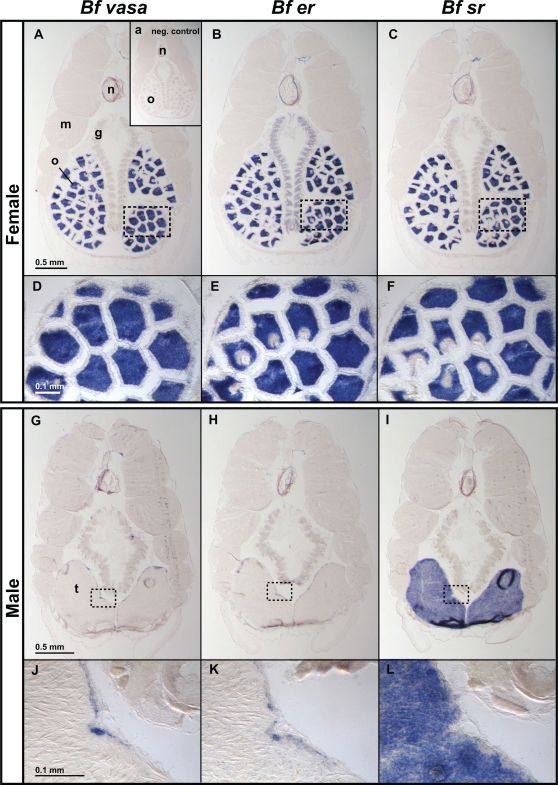
Gene duplication is the predominant mechanism for the evolution of new genes. Major existing models of this process assume that duplicate genes are redundant; degenerative mutations in one copy can therefore accumulate close to neutrally, usually leading to loss from the genome. When gene products dimerize or interact with other molecules for their functions, however, degenerative mutations in one copy may produce repressor alleles that inhibit the function of the other and are therefore exposed to selection. Here, we describe the evolution of a duplicate repressor by simple degenerative mutations in the steroid hormone receptors (SRs), a biologically crucial vertebrate gene family. We isolated and characterized the SRs of the cephalochordate Branchiostoma floridae, which diverged from other chordates just after duplication of the ancestral SR. The B. floridae genome contains two SRs: BfER, an ortholog of the vertebrate estrogen receptors, and BfSR, an ortholog of the vertebrate receptors for androgens, progestins, and corticosteroids. BfSR is specifically activated by estrogens and recognizes estrogen response elements (EREs) in DNA; BfER does not activate transcription in response to steroid hormones but binds EREs, where it competitively represses BfSR. The two genes are partially coexpressed, particularly in ovary and testis, suggesting an ancient role in germ cell development. These results corroborate previous findings that the ancestral steroid receptor was estrogen-sensitive and indicate that, after duplication, BfSR retained the ancestral function, while BfER evolved the capacity to negatively regulate BfSR. Either of two historical mutations that occurred during BfER evolution is sufficient to generate a competitive repressor. Our findings suggest that after duplication of genes whose functions depend on specific molecular interactions, high-probability degenerative mutations can yield novel functions, which are then exposed to positive or negative selection; in either case, the probability of neofunctionalization relative to gene loss is increased compared to existing models.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



References
-
- Orengo CA, Thornton JM. Protein families and their evolution-a structural perspective. Annu Rev Biochem. 2005;74:867–900. - PubMed
-
- Teichmann SA, Babu MM. Gene regulatory network growth by duplication. Nat Genet. 2004;36:492–496. - PubMed
-
- Haldane JBS. The part played by recurrent mutation in evolution. American Naturalist. 1933;67:5–19.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
