

MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA
- PMID: 18794355
- PMCID: PMC2573297
- DOI: 10.1128/MCB.00941-08
MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA
Abstract
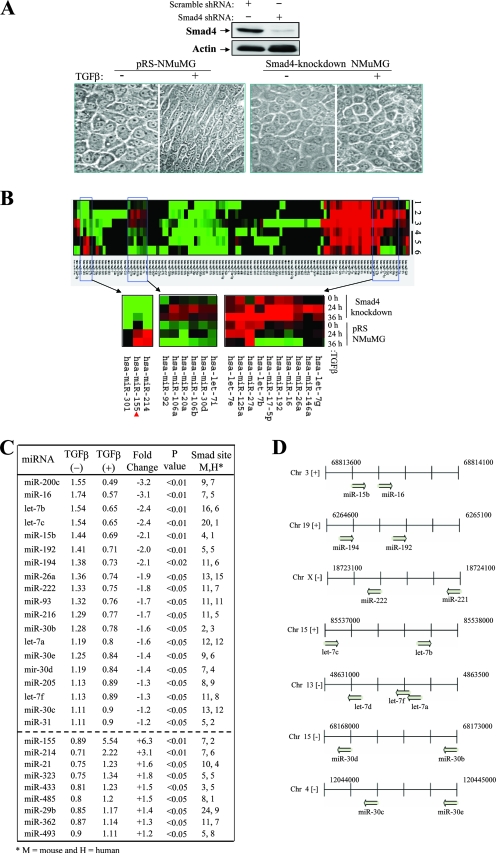
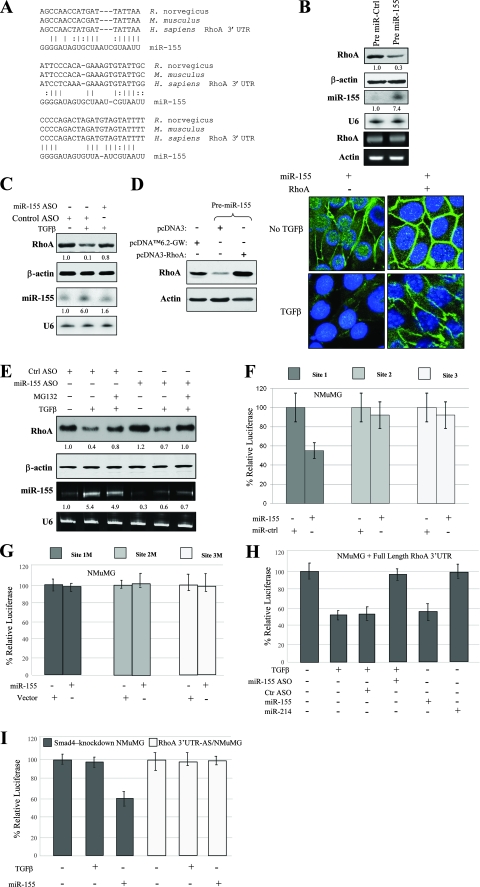
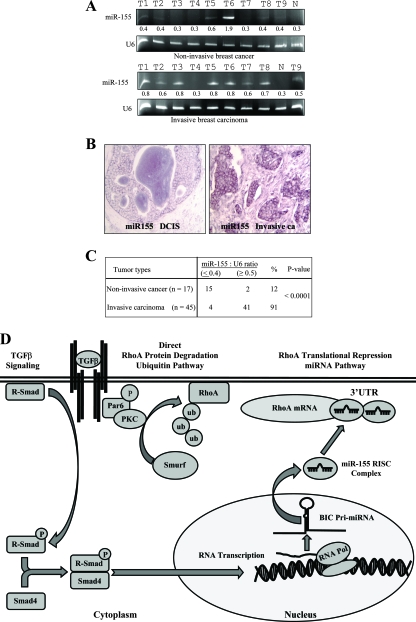
Transforming growth factor beta (TGF-beta) signaling facilitates metastasis in advanced malignancy. While a number of protein-encoding genes are known to be involved in this process, information on the role of microRNAs (miRNAs) in TGF-beta-induced cell migration and invasion is still limited. By hybridizing a 515-miRNA oligonucleotide-based microarray library, a total of 28 miRNAs were found to be significantly deregulated in TGF-beta-treated normal murine mammary gland (NMuMG) epithelial cells but not Smad4 knockdown NMuMG cells. Among upregulated miRNAs, miR-155 was the most significantly elevated miRNA. TGF-beta induces miR-155 expression and promoter activity through Smad4. The knockdown of miR-155 suppressed TGF-beta-induced epithelial-mesenchymal transition (EMT) and tight junction dissolution, as well as cell migration and invasion. Further, the ectopic expression of miR-155 reduced RhoA protein and disrupted tight junction formation. Reintroducing RhoA cDNA without the 3' untranslated region largely reversed the phenotype induced by miR-155 and TGF-beta. In addition, elevated levels of miR-155 were frequently detected in invasive breast cancer tissues. These data suggest that miR-155 may play an important role in TGF-beta-induced EMT and cell migration and invasion by targeting RhoA and indicate that it is a potential therapeutic target for breast cancer intervention.
Figures



References
-
- Ambros, V. 2004. The functions of animal microRNAs. Nature 431350-355. - PubMed
-
- Deckers, M., M. van Dinther, J. Buijs, I. Que, C. Lowik, G. van der Pluijm, and P. ten Dijke. 2006. The tumor suppressor Smad4 is required for transforming growth factor beta-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 662202-2209. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous