

Ligand-dependent ubiquitination of Smad3 is regulated by casein kinase 1 gamma 2, an inhibitor of TGF-beta signaling
- PMID: 18794808
- PMCID: PMC2643063
- DOI: 10.1038/onc.2008.337
Ligand-dependent ubiquitination of Smad3 is regulated by casein kinase 1 gamma 2, an inhibitor of TGF-beta signaling
Abstract
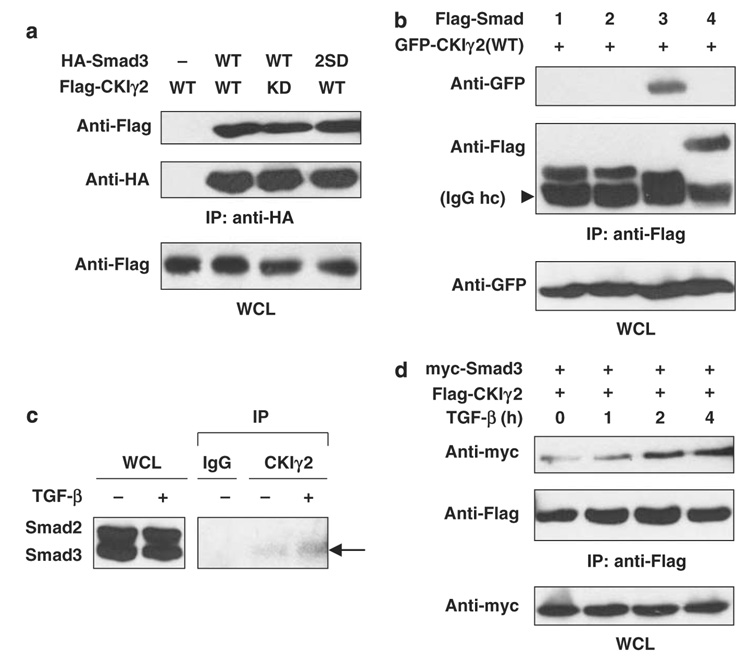
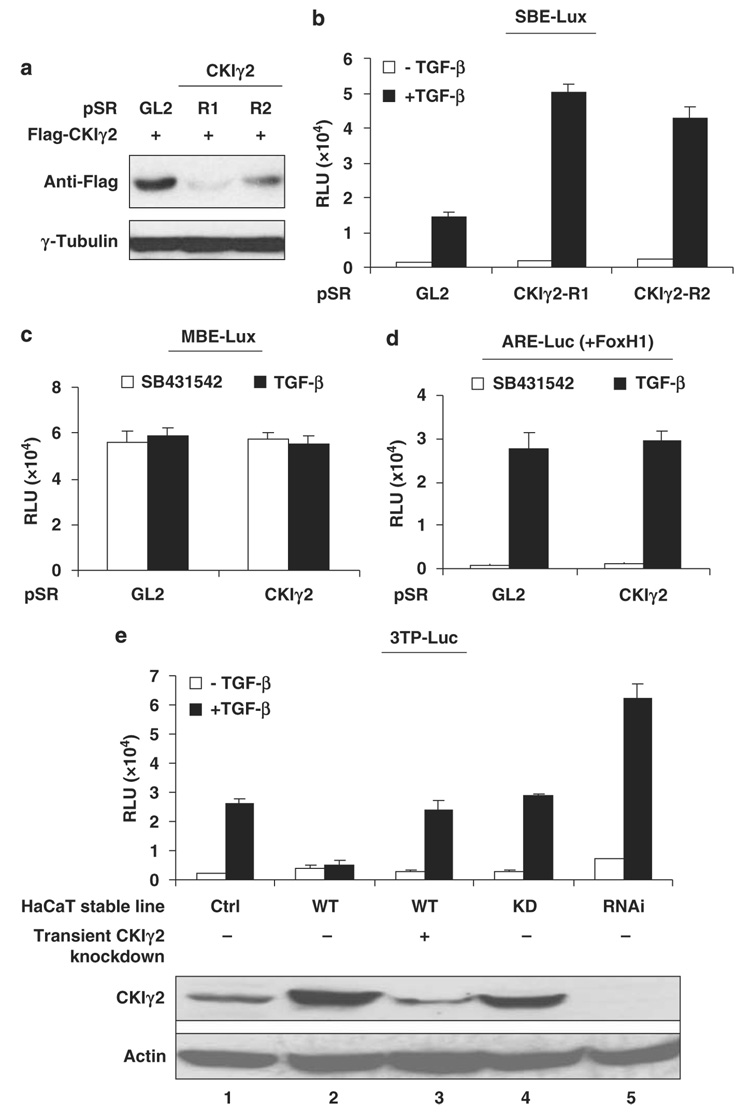
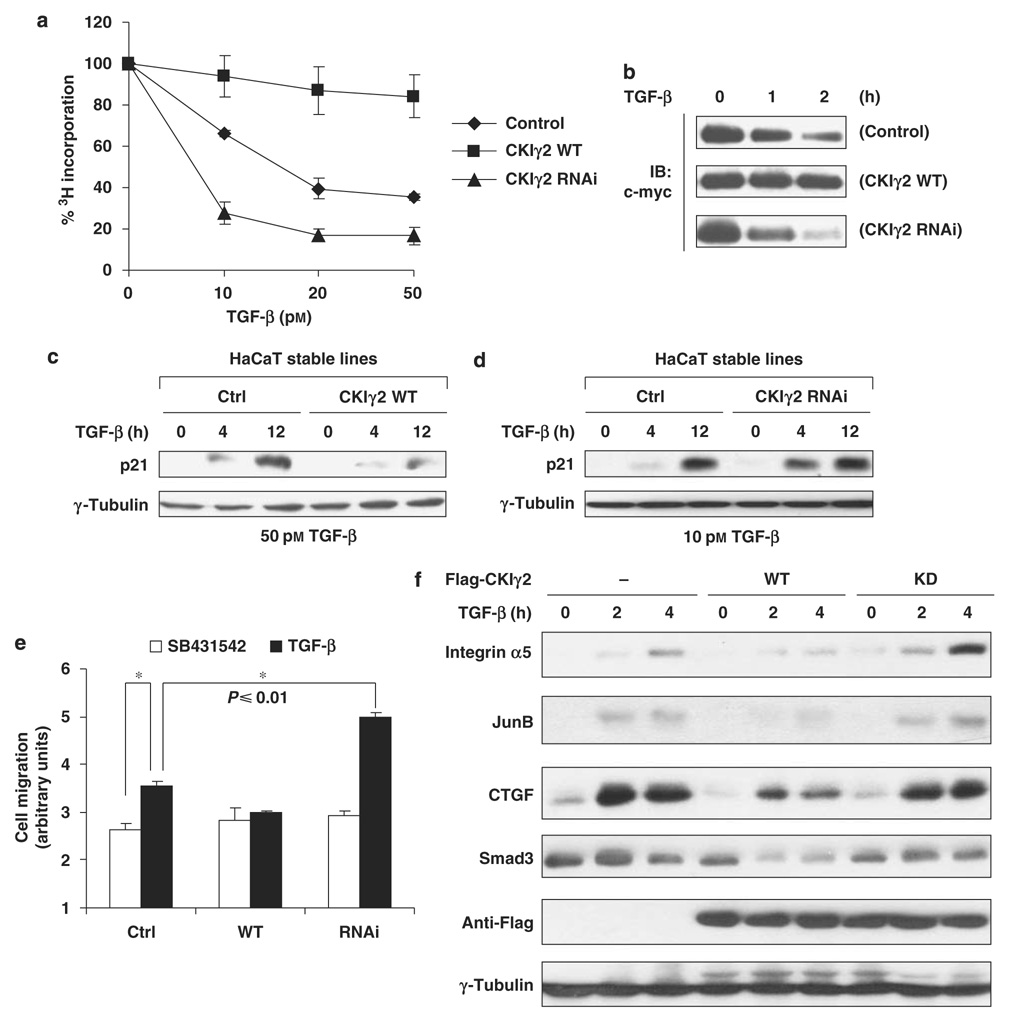
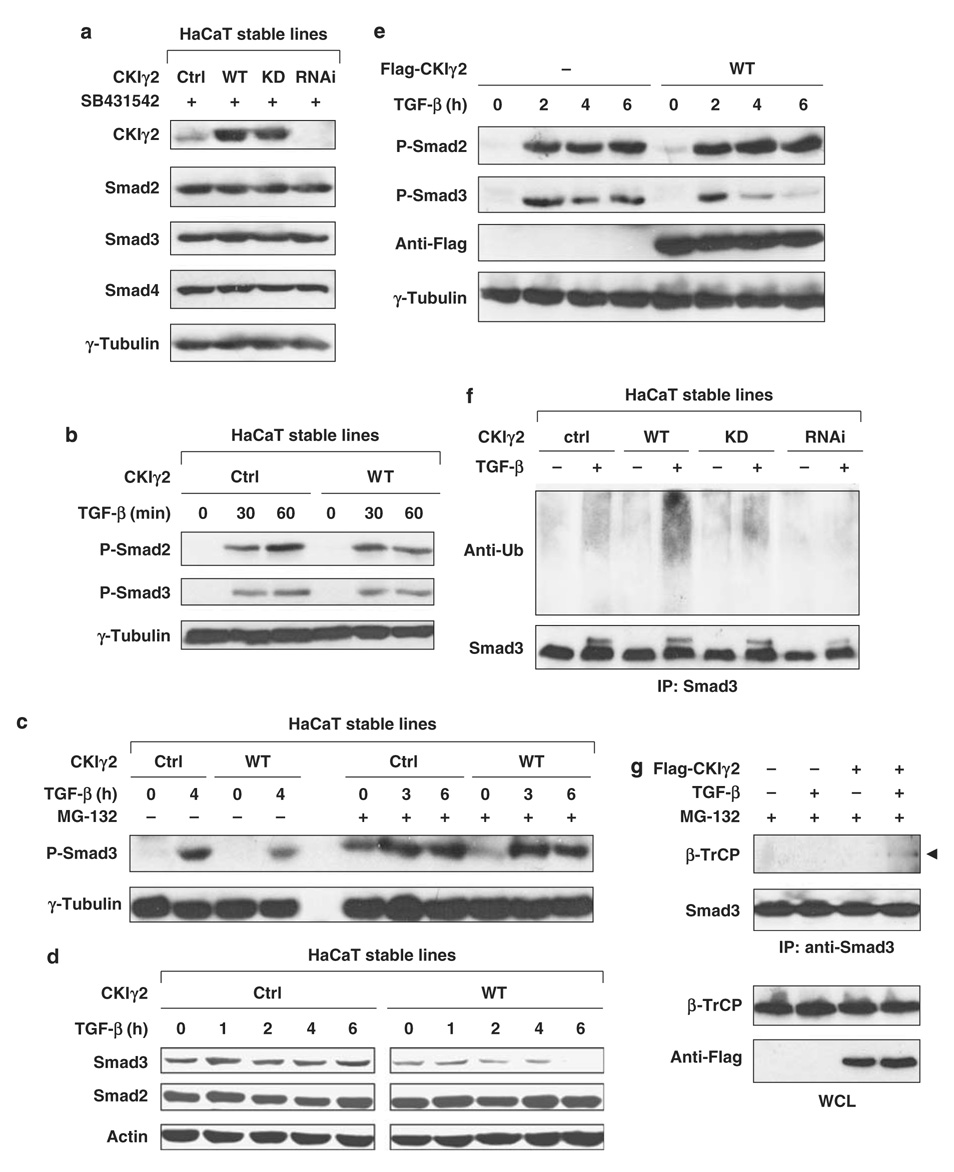
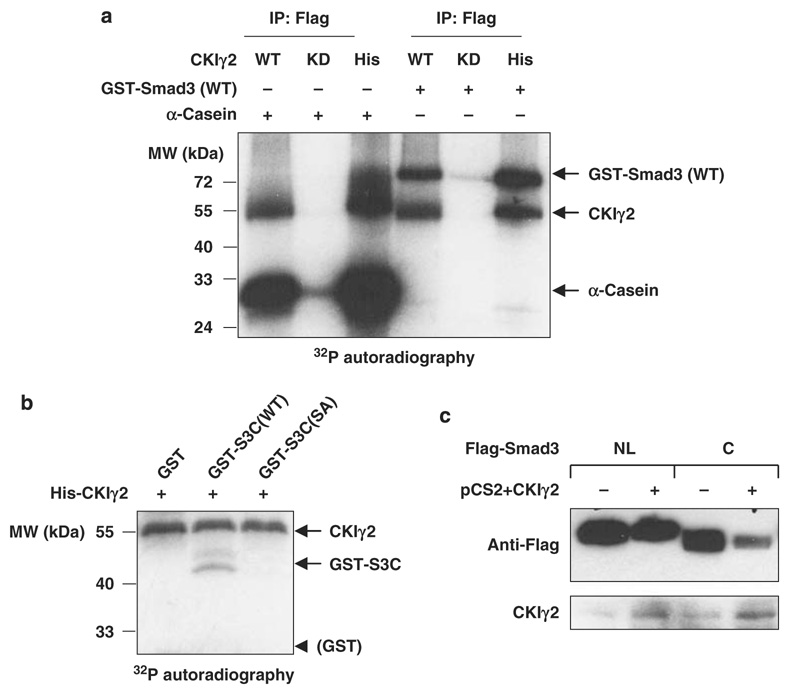
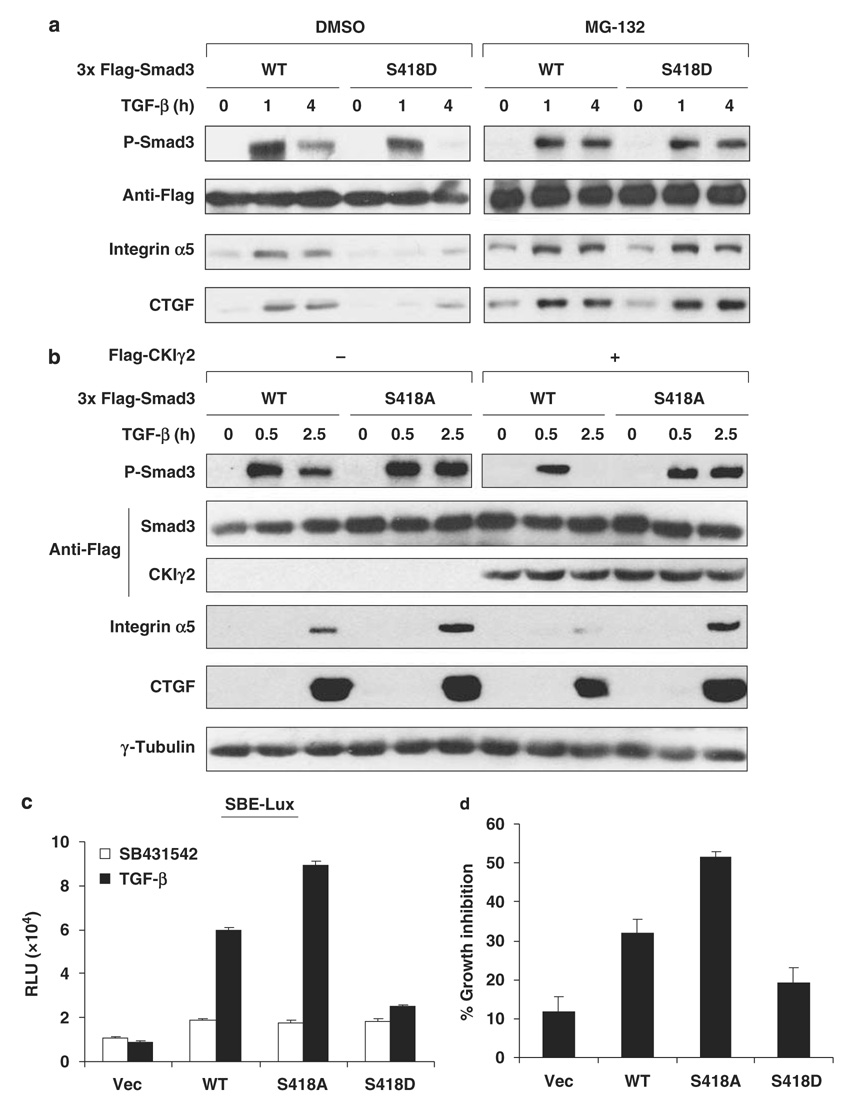
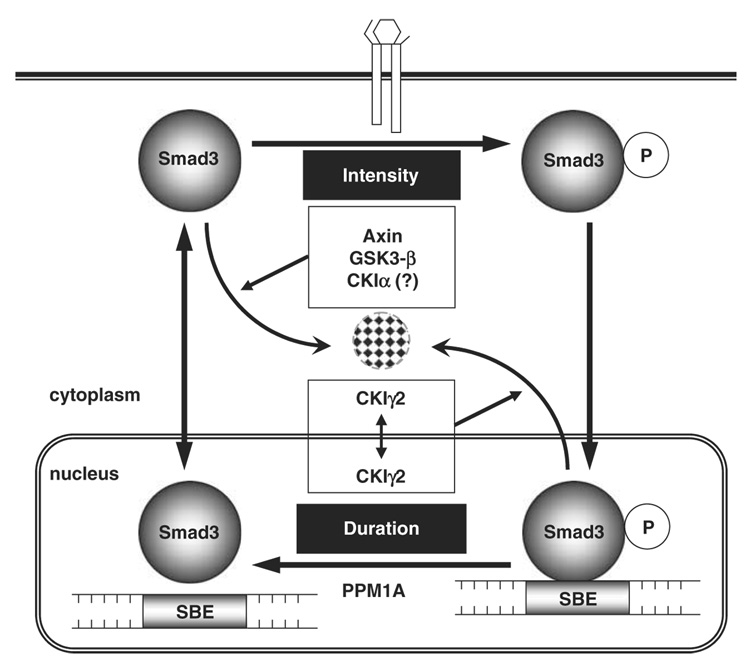
Transforming growth factor-beta (TGF-beta) elicits a variety of cellular activities primarily through a signaling cascade mediated by two key transcription factors, Smad2 and Smad3. Numerous regulatory mechanisms exist to control the activity of Smad3, thereby modulating the strength and specificity of TGF-beta responses. In search for potential regulators of Smad3 through a yeast two-hybrid screen, we identified casein kinase 1 gamma 2 (CKIgamma2) as a novel Smad3-interacting protein. In mammalian cells, CKIgamma2 selectively and constitutively binds Smad3 but not Smad1, -2 or -4. Functionally, CKIgamma2 inhibits Smad3-mediated TGF-beta responses including induction of target genes and cell growth arrest, and this inhibition is dependent on CKIgamma2 kinase activity. Mechanistically, CKIgamma2 does not affect the basal levels of Smad proteins or activity of the receptors. Rather, CKIgamma2 preferentially promotes the ubiquitination and degradation of activated Smad3 through direct phosphorylation of its MH2 domain at Ser418. Importantly, mutation of Ser418 to alanine or aspartic acid causes an increase or decrease of Smad3 activity, respectively, in the presence of TGF-beta. CKIgamma2 is the first kinase known to mark activated Smad3 for destruction. Given its negative function in TGF-beta signaling and its reported overexpression in human cancers, CKIgamma2 may act as an oncoprotein during tumorigenesis.
Figures
References
-
- Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–520. - PubMed
-
- Davidson G, Wu W, Shen J, Bilic J, Fenger U, Stannek P, et al. Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature. 2005;438:867–872. - PubMed
-
- Dennler S, Huet S, Gauthier JM. A short amino-acid sequence in MH1 domain is responsible for functional differences between Smad2 and Smad3. Oncogene. 1999;18:1643–1648. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
