

The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK
- PMID: 18796143
- PMCID: PMC2553763
- DOI: 10.1186/1750-1172-3-24
The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK
Abstract
Background: Mucopolysaccharidosis type I (MPS I) is a rare lysosomal storage disease subdivided into three phenotypes of increasing severity: Scheie, Hurler-Scheie and Hurler. To gauge the effectiveness of treatments and to determine the load likely to fall on health-care systems, it is necessary to understand the prevalence and natural progression of the disease especially with regard to life-expectancy. In general such data on the natural history of lysosomal storage diseases is sparse.
Methods: Analysis of prevalence and patient survival in MPS I disease using a unique longitudinal data set initiated and maintained over a period of more than 20 years by the Society for Mucopolysaccharide Diseases (UK).
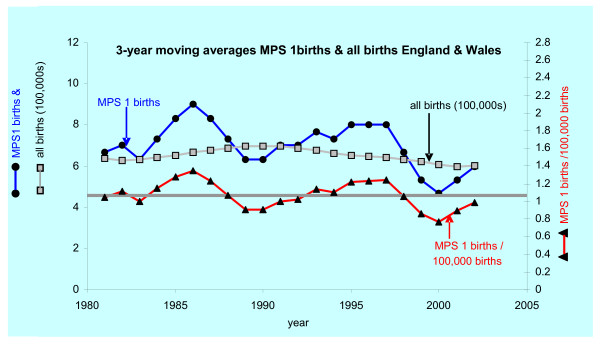
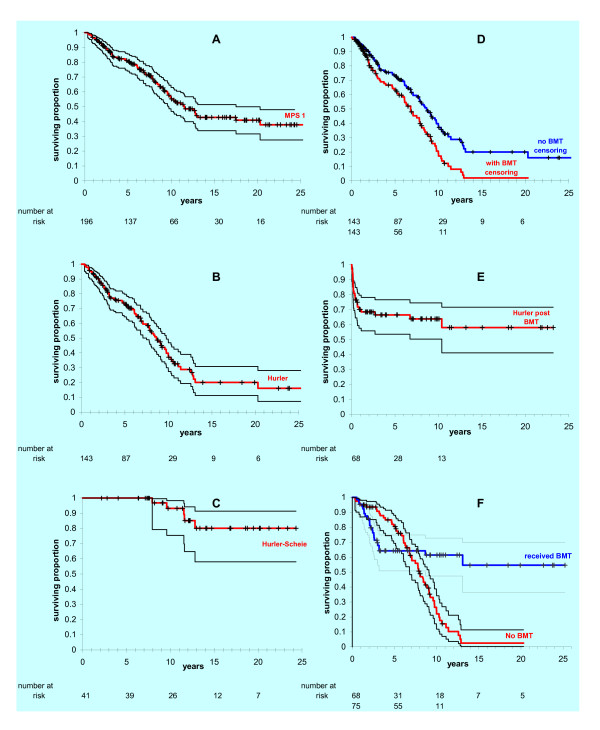
Results: The birth prevalence of MPS I in England and Wales over the period 1981 to 2003 was 1.07/100,000 births and within +/- 5% of estimates reported in several studies that examined reasonably large populations. The median survival for MPS I patients (including all phenotypes irrespective of various treatments) was found by Kaplan-Meier analysis to be 11.6 years. This result was driven by the relatively poor survival of patients with the Hurler phenotype who, irrespective of any treatments received, had a median survival of 8.7 years; when censoring for receipt of bone marrow transplant (BMT) was implemented median survival of Hurler patients was diminished to 6.8 years. The difference between these survival curves was statistically significant by log rank test and can be attributed to beneficial effects of BMT and or selection of patients with superior prognosis for intervention with BMT. Survival curves for Hurler patients who received and did not receive BMT were very different. Probability of survival at 2 year after BMT was ~68% and was similar to this after 5 years (66%) and ten years (64%); the mean age of Hurler patients at receipt of BMT was 1.33 years (range 0.1 to 3 years). Follow up was insufficient to determine median survival of the milder phenotypes however, unsurprisingly, this was clearly superior to that for Hurler patients.
Conclusion: The birth prevalence of MPS I in England and Wales is 1.07/100,000 and the median survival for MPS I patients is 11.6 years.
Figures
References
-
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Sly WS, editor. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2000. pp. 3421–3452.
-
- Wraith JE, Beck M, Lane R, van der PA, Shapiro E, Xue Y, et al. Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: results of a multinational study of recombinant human alpha-L-iduronidase (laronidase) Pediatrics. 2007;120:e37–e46. doi: 10.1542/peds.2006-2156. - DOI - PubMed
-
- Connock M, Burls AB, Frew E, Fry-Smith A, Juarez-Garcia A, McCabe C, et al. The clinical effectiveness and cost-effectiveness of enzyme replacement therapy for Gaucher's disease. Health Technol Assess. 2006;10 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
