

The impact of peptide abundance and dynamic range on stable-isotope-based quantitative proteomic analyses
- PMID: 18798661
- PMCID: PMC2746028
- DOI: 10.1021/pr800333e
The impact of peptide abundance and dynamic range on stable-isotope-based quantitative proteomic analyses
Abstract
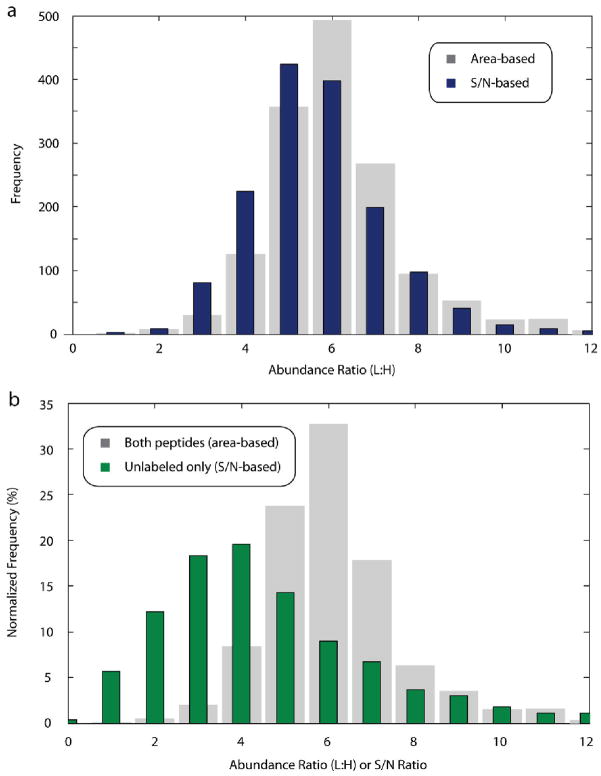
Recently, mass spectrometry has been employed in many studies to provide unbiased, reproducible, and quantitative protein abundance information on a proteome-wide scale. However, how instruments' limited dynamic ranges impact the accuracy of such measurements has remained largely unexplored, especially in the context of complex mixtures. Here, we examined the distribution of peptide signal versus background noise (S/N) and its correlation with quantitative accuracy. With the use of metabolically labeled Jurkat cell lysate, over half of all confidently identified peptides had S/N ratios less than 10 when examined using both hybrid linear ion trap-Fourier transform ion cyclotron resonance and Orbitrap mass spectrometers. Quantification accuracy was also highly correlated with S/N. We developed a mass precision algorithm that significantly reduced measurement variance at low S/N beyond the use of highly accurate mass information alone and expanded it into a new software suite, Vista. We also evaluated the interplay between mass measurement accuracy and S/N; finding a balance between both parameters produced the greatest identification and quantification rates. Finally, we demonstrate that S/N can be a useful surrogate for relative abundance ratios when only a single species is detected.
Figures
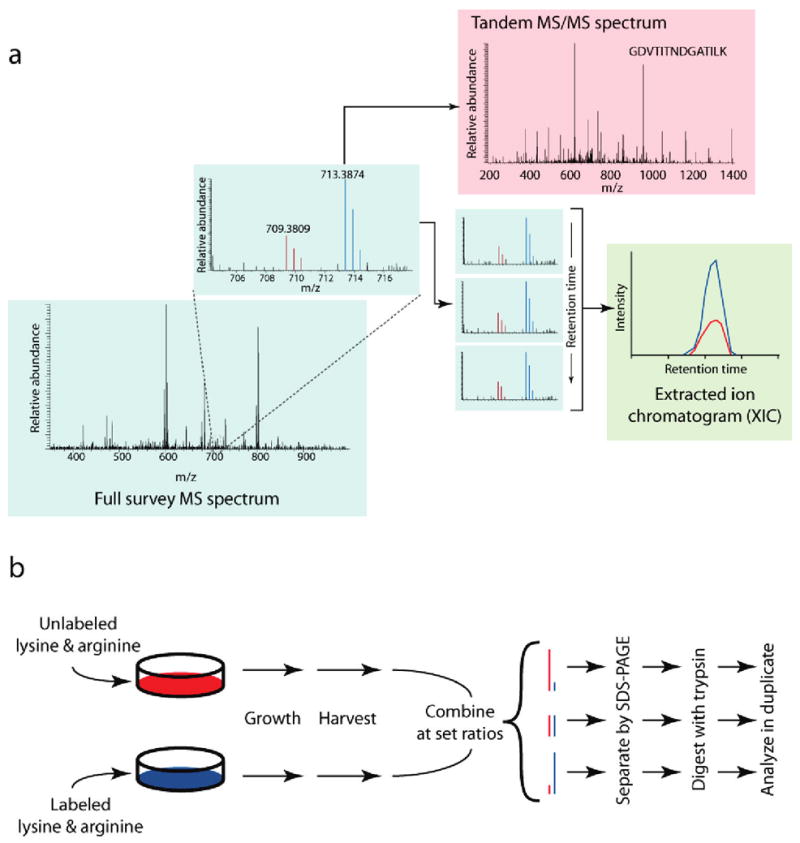
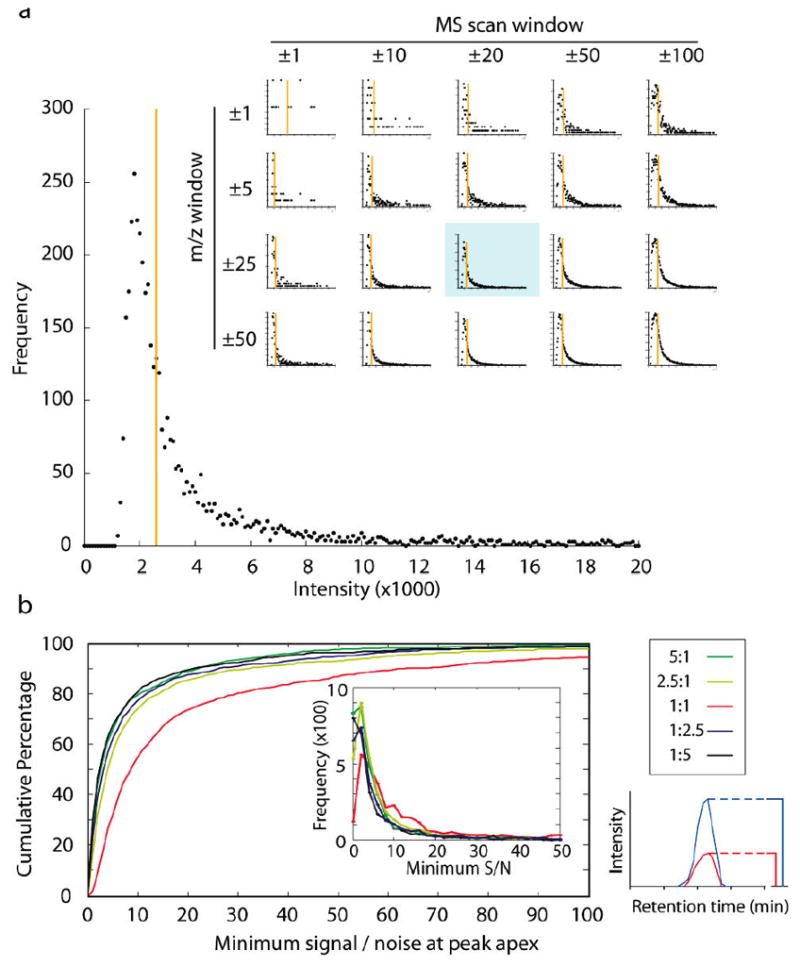
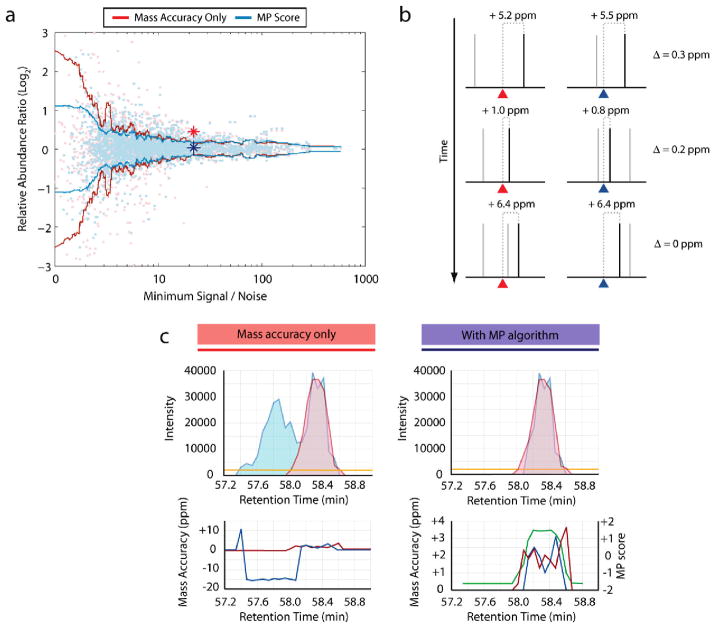
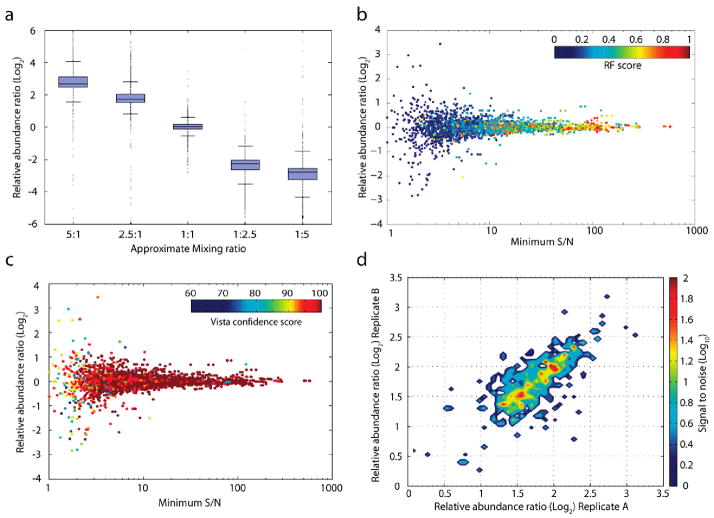
References
-
- Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. - PubMed
-
- Dengjel J, et al. Quantitative proteomic assessment of very early cellular signaling events. Nat Biotechnol. 2007;25:566–568. - PubMed
-
- Dong MQ, et al. Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science. 2007;317:660–663. - PubMed
-
- Ong SE, Foster LJ, Mann M. Mass spectrometric-based approaches in quantitative proteomics. Methods. 2003;29:124–130. - PubMed
-
- Gygi SP, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
