

doi: 10.1038/nature07368.
Computational prediction of small-molecule catalysts
Affiliations
- PMID: 18800129
- PMCID: PMC2717898
- DOI: 10.1038/nature07368
Item in Clipboard
Computational prediction of small-molecule catalysts
Nature.
.
Abstract
Most organic and organometallic catalysts have been discovered through serendipity or trial and error, rather than by rational design. Computational methods, however, are rapidly becoming a versatile tool for understanding and predicting the roles of such catalysts in asymmetric reactions. Such methods should now be regarded as a first line of attack in the design of catalysts.
Figures
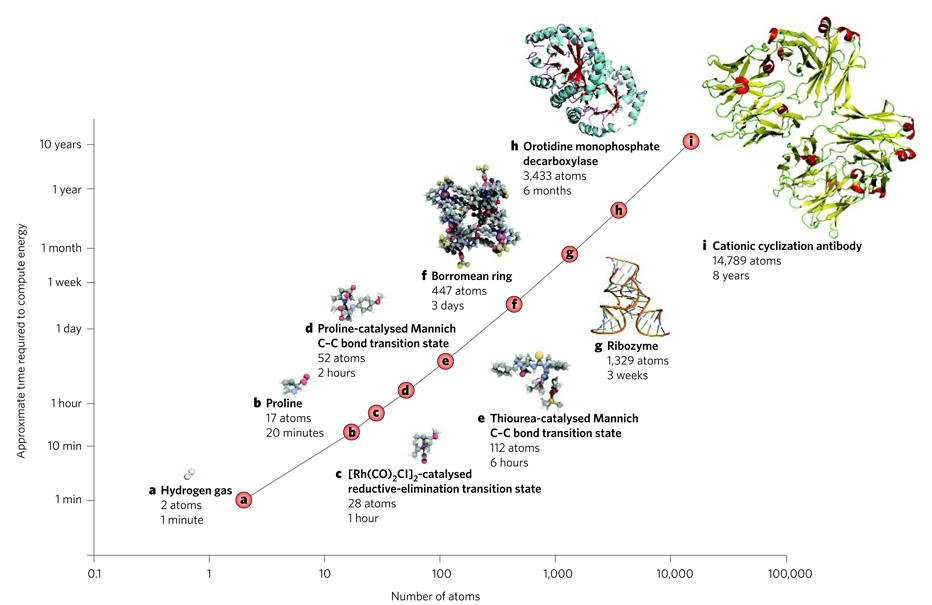
Shown is the approximate time taken to compute the structure of molecules of various sizes by using density functional theory and double-zeta basis sets on a modern desktop computer with appropriate memory and storage. The time required (shown on a log scale) increases exponentially with the size of the molecule. Structures were generated using PyMOL, QuteMol or CYLview.
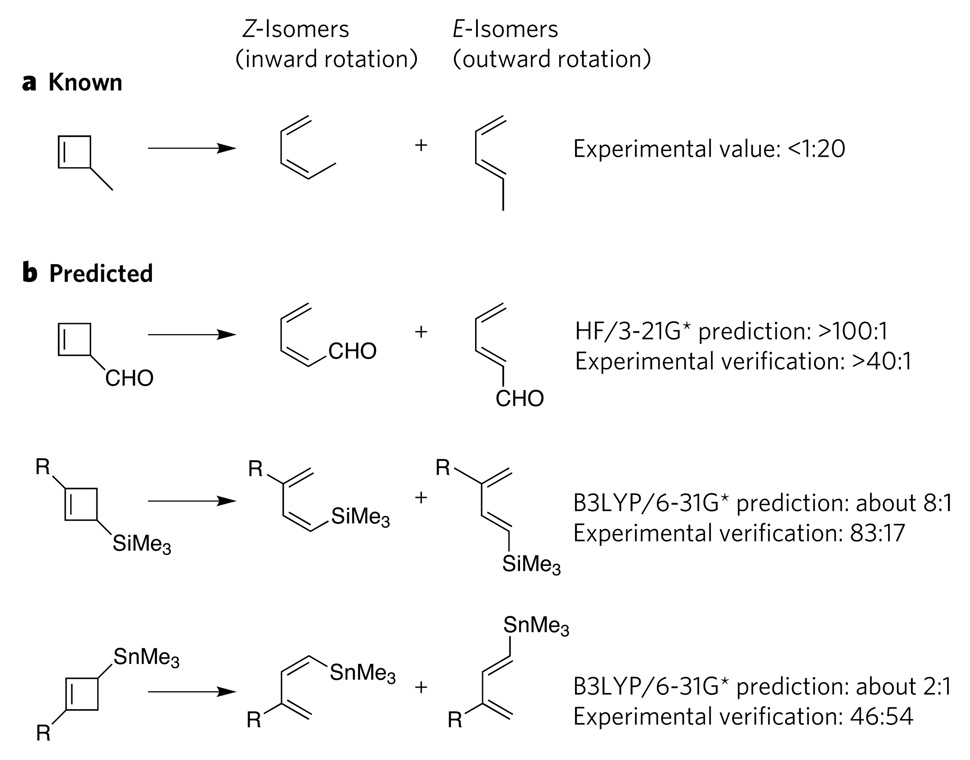
a, In cyclobutene ring-opening reactions, 3-methylcyclobutene forms mainly the E-isomer of 1-methylbutadiene (as well as a small amount of the Z-isomer); the experimentally determined ratio is <1 Z-isomer to 20 E-isomers. b, Using this knowledge as a starting point, computations correctly predicted the stereochemical outcomes of three analogous reactions–, as verified by subsequent experiments, even though the products were mainly Z-isomers rather than E-isomers. In these reactions, the electron-withdrawing substituents favour inward rotation, whereas, in the original reaction, the electron-donating substituent favours outward rotation. HF/3-21G* is an ab initio computational method, and B3LYP/6-31G* is a density-functional-theory computational method. Me, methyl; R, CMe2Ph.
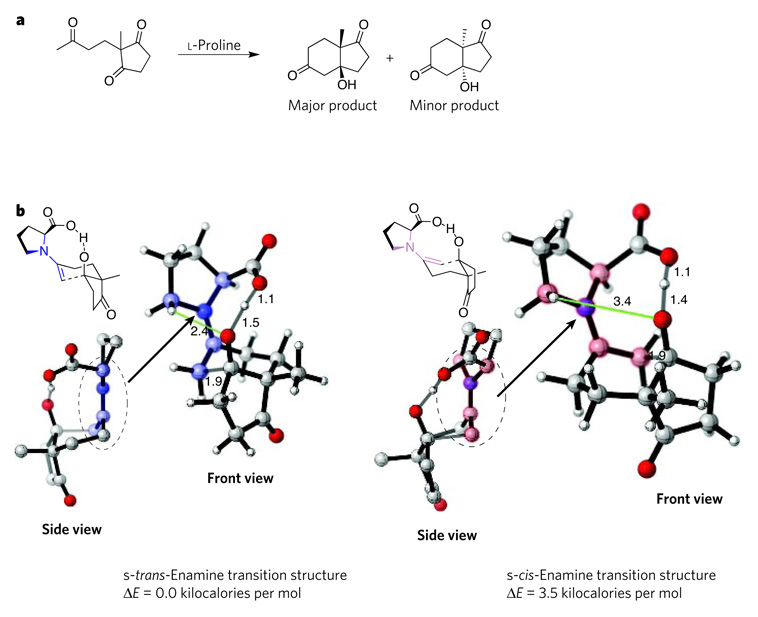
The Hajos–Parrish reaction (a) was one of the first reported organocatalytic processes. Four possible transition states for this reaction were modelled using the hybrid density-functionaltheory method B3LYP and the basis set 6-31G*. The computations revealed that the most favoured transition state involves the s-trans-enamine intermediate (b, left), which incorporates a shorter, and therefore more strongly stabilizing, electrostatic interaction (green) between a hydrogen atom in the pyrrolidine ring and the oxygen atom of the developing alkoxide than does the s-cis-enamine intermediate (b, right). The side view of the s-trans enamine shows that this intermediate is a stable, undistorted (planar) developing iminium ion, whereas the s-cis-enamine intermediate is an unstable, distorted (non-planar) developing iminium ion. The favoured transition state leads to the formation of the major product that is observed experimentally, whereas the other transition state leads to the minor product. The main structural differences between the two intermediates are shaded in pale blue (s-trans form) and pale pink (s-cis form). Carbon is shown in grey, hydrogen in white, oxygen in red and nitrogen in blue. Distances between atoms are indicated in angstroms. Structures were generated using CYLview. E, energy.
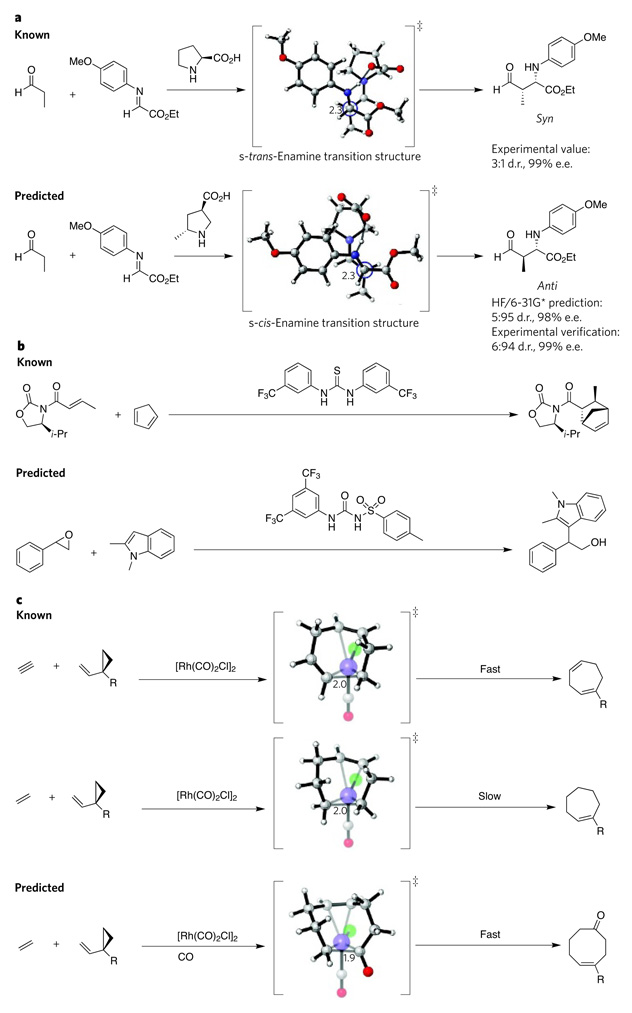
a, Most Mannich reactions catalysed by organocatalysts mainly yield products in the syn configuration (top). Chemists designed an organocatalyst selective for the anti configuration (bottom) based on computational analysis of information from a proline-catalysed Mannich reaction. b, The design of an epoxide-activating urea catalyst (bottom) came from the computational analysis of known thiourea catalysts (top) that activate carbonyl-containing compounds. c, Rhodium-catalysed [5 + 2]-cycloaddition reactions of vinylcyclopropanes with allenes (not shown) or alkynes (top) are fast, but the analogous reactions with alkenes (centre) are slow. Computational analysis of these reactions led to the design of an analogous [5 + 2 + 1]-multicomponent-cycloaddition reaction that yields cyclooctenones (bottom). Carbon is shown in grey, hydrogen in white, oxygen in red, nitrogen in blue, chloride in green and rhodium in purple (large spheres). Critical carbon–carbon bond-forming distances in the transition structure are indicated in angstroms. Transition-state structures were generated using CYLview. d.r., diastereomeric ratio; e.e., enantiomeric excess; Et, ethyl; i-Pr, isopropyl; R, alkoxy, alkyl, siloxy; ‡, transition state.
References
-
- Gaussian 03, Revision C.02. Wallingford, Connecticut: Gaussian; 2004.
-
- Zhao Y, Truhlar DG. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008;2:157–167. - PubMed
-
- Cramer CJ. Essentials of Computational Chemistry: Theories and Models 2nd edn. Wiley; 2004.
-
- Weik MH. A Third Survey of Domestic Electronic Digital Computing Systems. Maryland: Ballistic Research Laboratories, Aberdeen Proving Ground; 1961. pp. 526–536. Report No. 1115.
-
- Rudolf K, Spellmeyer DC, Houk KN. Prediction and experimental verification of the stereoselective electrocyclization of 3-formylcyclobutene. J. Org. Chem. 1987;52:3708–3710.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
