

Carbon-heteroatom bond formation catalysed by organometallic complexes
- PMID: 18800130
- PMCID: PMC2819340
- DOI: 10.1038/nature07369
Carbon-heteroatom bond formation catalysed by organometallic complexes
Abstract
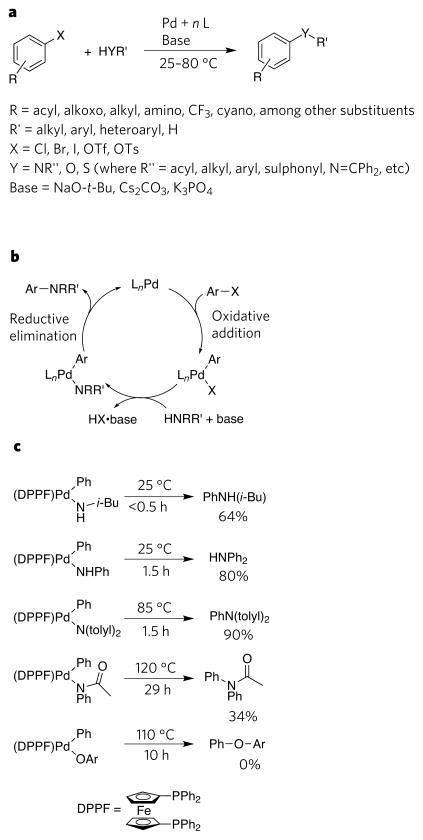
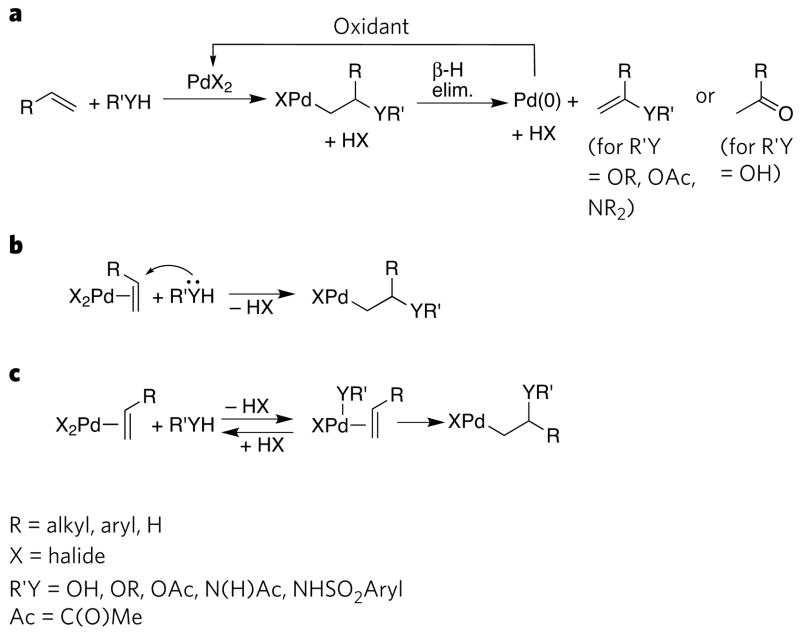
At one time the synthetic chemist's last resort, reactions catalysed by transition metals are now the preferred method for synthesizing many types of organic molecule. A recent success in this type of catalysis is the discovery of reactions that form bonds between carbon and heteroatoms (such as nitrogen, oxygen, sulphur, silicon and boron) via complexes of transition metals with amides, alkoxides, thiolates, silyl groups or boryl groups. The development of these catalytic processes has been supported by the discovery of new elementary reactions that occur at metal-heteroatom bonds and by the identification of factors that control these reactions. Together, these findings have led to new synthetic processes that are in daily use and have formed a foundation for the development of processes that are likely to be central to synthetic chemistry in the future.
Figures







References
-
- Muci AR, Buchwald SL. Practical palladium catalysts for C–N and C–O bond formation. Top Curr Chem. 2002;219:131–209. This paper reviews the primary literature on cross-coupling to form C–N and C–O bonds.
-
- Hartwig JF. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi EI, editor. Vol. 1. Wiley-Interscience; 2002. pp. 1051–1096.
-
- Hartwig JF. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi EI, editor. Vol. 1. Wiley-Interscience; 2002. pp. 1097–1106.
-
- Dick AR, Sanford MS. Transition metal catalyzed oxidative functionalization of carbon–hydrogen bonds. Tetrahedron. 2006;62:2439–2463. This paper reviews various modern approaches to C–H bond functionalization to form C–heteroatom bonds.
-
- Hull KL, Anani WQ, Sanford MS. Palladium-catalyzed fluorination of carbon–hydrogen bonds. J Am Chem Soc. 2006;128:7134–7135. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
