

Review
doi: 10.1038/nature07370.
Natural products as inspiration for the development of asymmetric catalysis
Affiliations
- PMID: 18800131
- PMCID: PMC2562237
- DOI: 10.1038/nature07370
Item in Clipboard
Review
Natural products as inspiration for the development of asymmetric catalysis
Nature.
.
Abstract
Biologically active natural products often contain particularly challenging structural features and functionalities in terms of synthesis. Perhaps the greatest difficulties are those caused by issues of stereochemistry. A useful strategy for synthesizing such molecules is to devise methods of bond formation that provide opportunities for using enantioselective catalysis. In using this tactic, the desire for a particular target structure ultimately drives the development of catalytic methods. New enantioselective catalytic methods contribute to a greater fundamental understanding of how bonds can be constructed and lead to valuable synthetic technologies that are useful for a variety of applications.
Figures
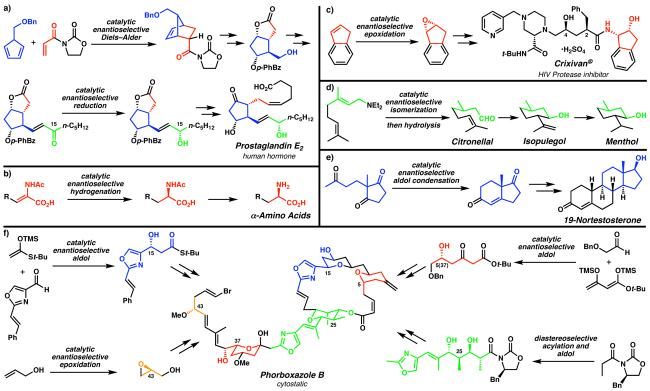
Natural products and pharmaceuticals often possess challenging structural features and functionalities, particularly stereochemical elements. (a) Corey's synthesis of prostaglandin E2 utilizing an asymmetric Diels–Alder reaction and carbonyl reduction. Various industrial applications, including (b) the asymmetric hydrogenation toward α-amino acid building blocks, (c) the asymmetric epoxidation toward Merck's HIV protease inhibitor Crixivan®, and (d) the asymmetric isomerization of allylic amines en route to commodity chemicals, have explored the limits of catalysis and efficiency to minimize cost. The asymmetric construction of C–C bonds, apparent in (e) the amino acid-catalyzed intramolecular aldol condensation toward important steroid building blocks, and (f) various enantioselecitive intermolecular aldol reactions toward fragments of the cytostatic agent phorboxazole B, maximize the use of stereochemical information of precious intermediates through convergency. (Reduced to 45%)

The rapid and selective synthesis of β-enamino amide 3 enabled a key rhodium/5-catalyzed enantioselective hydrogenation under mildly acidic conditions to directly reveal the β-amino amide in Merck's synthesis of Januvia®, an FDA-approved treatment for type II diabetes., DMAP, 4-(N,N-dimethylamino)pyridine; DMA, N,N-dimethylacetamide; PivCl, pivaloyl chloride; COD, cyclooctadiene. (Reduced to 45%)
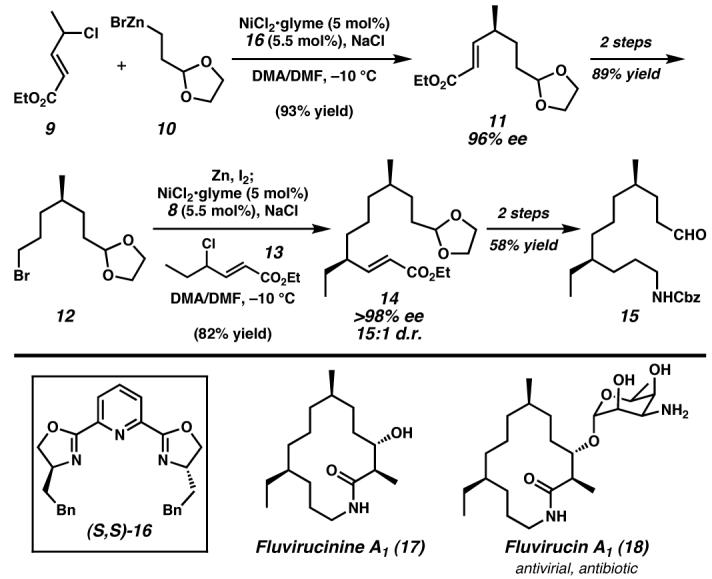
Sequential asymmetric C(sp3)–C(sp3) Negishi cross-couplings of racemic allylic chlorides and alkylzinc reagents catalyzed by nickel/(S,S)-16 enabled the rapid formal synthesis of Fluvirucinine A1 with excellent enantio- and diastereoselectivity, highlighting a creative solution to remote stereochemical control in unfunctionalized systems. Glyme, 1,2-dimethoxyethane; DMF, N,N-dimethylformamide. (Reduced to 45%)

The palladium/23-catalyzed enantioselective intramolecular Heck cyclization of 19 forged the C(9a) all-carbon quaternary stereocenter, and upon acid-mediated cyclization, the polycyclic core of minfiensine. The application of this method allowed the diastereoselective preparation of the remaining stereocenter and completion of the alkaloid. PMP, 1,2,2,6,6-pentamethylpiperidine. (Reduced to 45%)
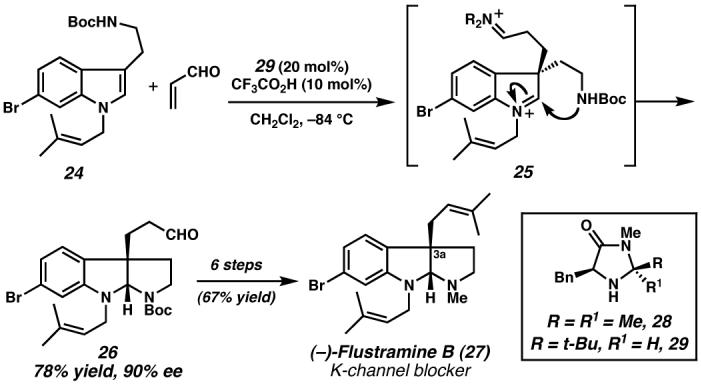
The enantioselective C(3a) alkylation of tryptamine derivative 24 catalyzed by amine 29, upon subsequent intramolecular cyclization, provided the pyrroloindoline core of (−)-flustramine B. This selective cascade process prepared the all-carbon quaternary stereocenter rapidly and efficiently enabling completion of this K-channel blocker. (Reduced to 45%)

Facile prepartion of hydroxylactam 31 facilitated the asymmetric Pictet–Spengler cyclization catalyzed by thiourea 32. The enantioselective N-acyl iminium ion cyclization enabled rapid construction of the alkaloid (+)-harmicine following this key transformation. TMSCl, chlorotrimethylsilane; THF, tetrahydrofuran. (Reduced to 45%)
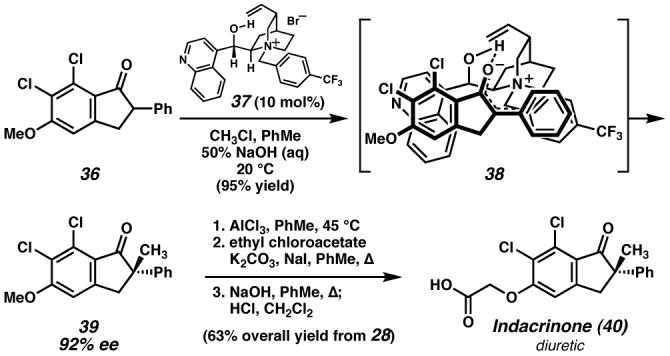
Merck scientists developed an enantioselective alkaloid salt-catalyzed phase-transfer enolate alkylation to gain access to indanone 39 with excellent stereoselectivity en route to the diuretic drug candidate indacrinone. (Reduced to 45%)
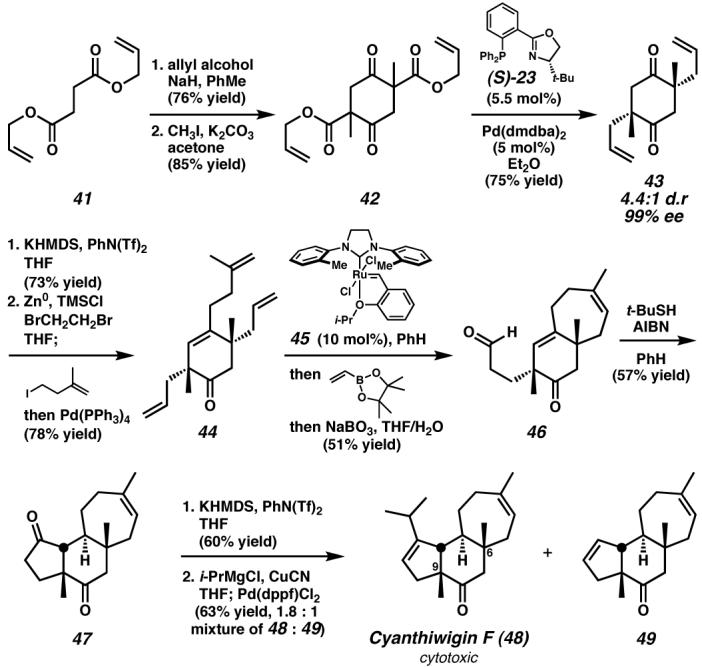
Preparation of a diastereomeric mixture of racemic and meso β-ketoester 42 allowed the strategic application of the palladium/(S)-23-catalyzed double enantioselective enolate alkylation to generate both all-carbon quaternary stereocenters of 43 with excellent stereoselectivity. A subsequent Negishi coupling, ring-closing metathesis, and radical-mediated aldehyde-olefin cyclization rapidly constructed the tricyclic core of cyanthiwigin F, completing a synthesis highlighted by several steps that involve the generation of multiple C–C bonds. dmdba, bis(3,5-dimethyoxybenzylidene)acetone; KHMDS, potassium bis(trimethylsilyl)amide; PhN(Tf)2, phenyl bis(trifluoromethane)sulfonimide; AIBN, 2,2′-azobis(isobutyronitrile); dppf, 1,1′-bis(diphenylphosphino)ferrocene. (Reduced to 45%)
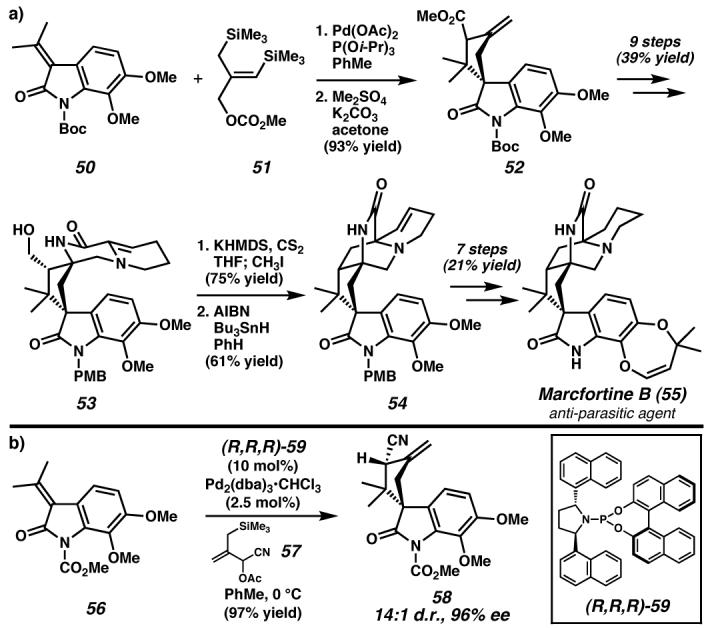
(a) The palladium-catalyzed trimethylenemetane [3 + 2] cyclization of oxindole 50 prepared cyclopentane 52 in excellent yield. Elaboration to tetracycle 53 facilitated an intramolecular radical cyclization to synthesize the core of marcfortine B, which was completed in seven additional steps. (b), The recent development of enantioselective palladium-catalyzed TMM cyclizations with (R,R,R)-59 allows access to spiro-annulated products such as 58 with high diastereo- and enantioselecitivity. The potential application to marcfortine B has not yet been realized. Boc, tert-butoxycarbonyl; dba, bis(benzylidene)acetone. (Reduced to 45%)
References
-
- Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. Wiley; New York: 1995. pp. 1–91.
-
- Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis, Vol. I–III. Springer-Verlag; Berlin: 2000.
-
- Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis, Supplement 1 & 2. Springer-Verlag; Berlin: 2004.
-
- Hoveyda AH. Asymmetric Catalysis in Target-Oriented Synthesis. In: Vögtle F, Stoddart JF, Shibasaki M, editors. Stimulating Concepts in Chemistry. Wiley-VCH; Weinheim: 2000. pp. 145–160.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
