

Myocardial ischaemia inhibits mitochondrial metabolism of 4-hydroxy-trans-2-nonenal
- PMID: 18800966
- PMCID: PMC3463957
- DOI: 10.1042/BJ20081615
Myocardial ischaemia inhibits mitochondrial metabolism of 4-hydroxy-trans-2-nonenal
Abstract
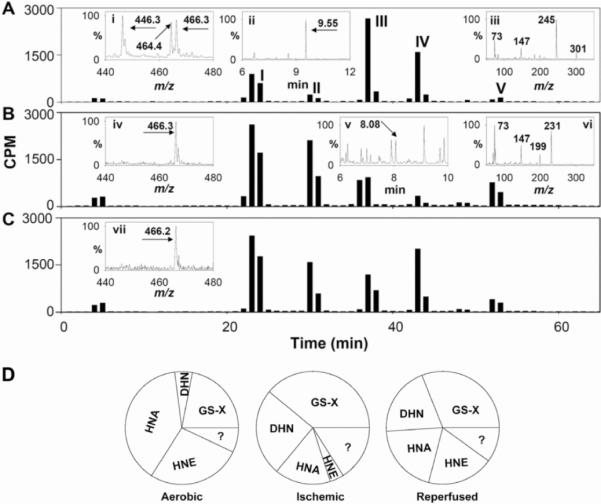
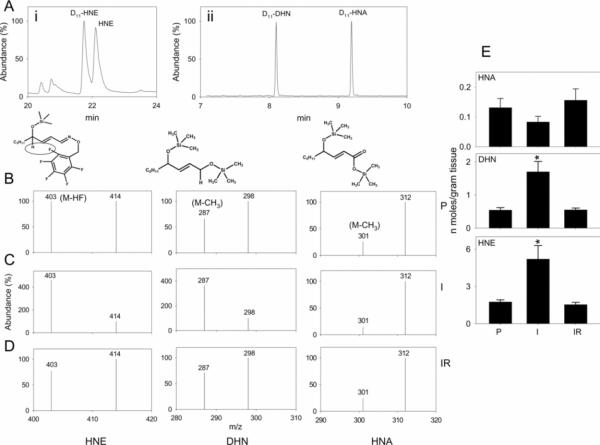
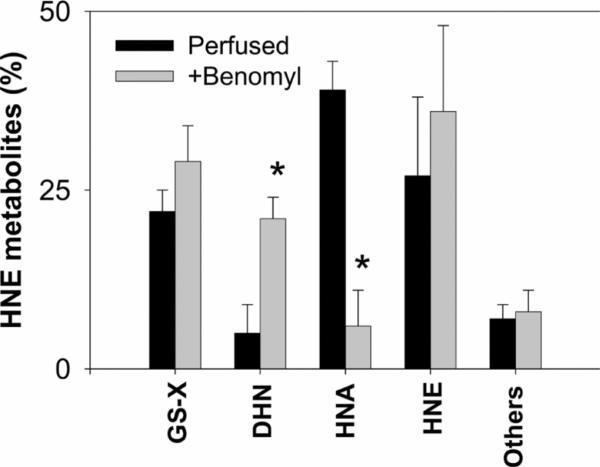
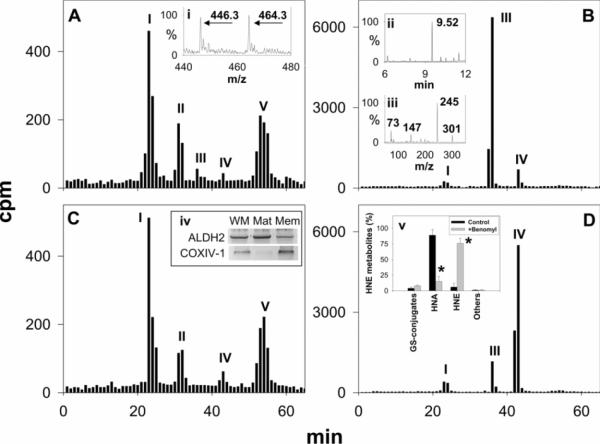
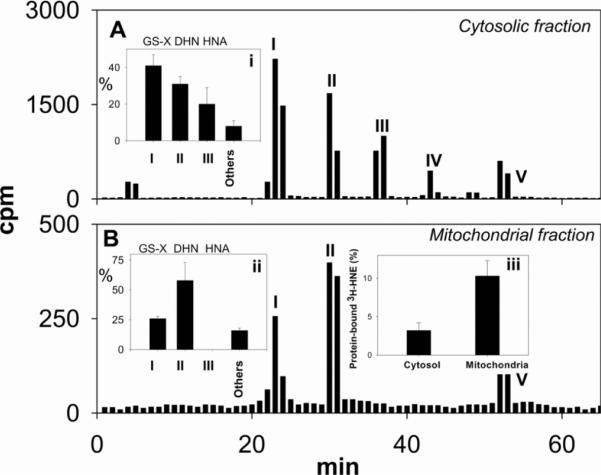
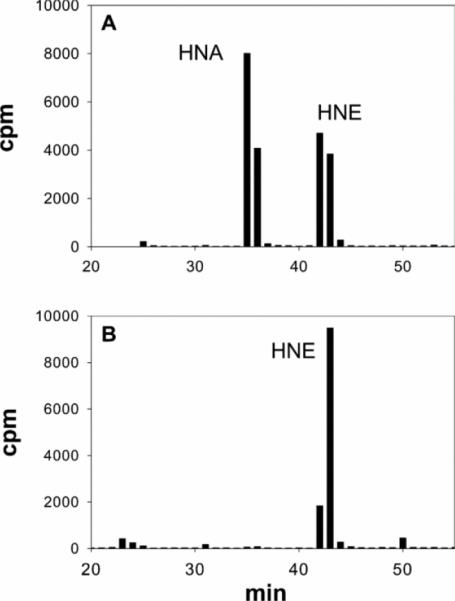
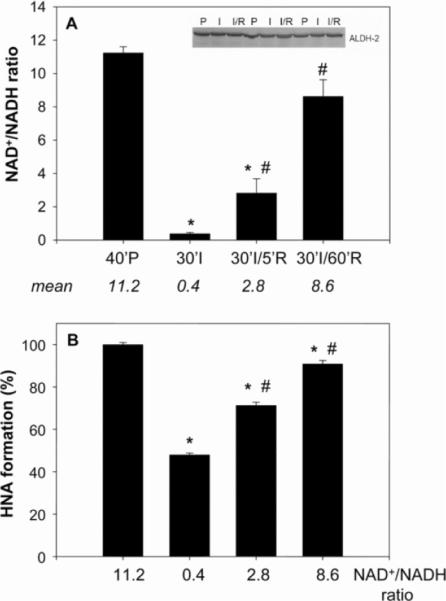
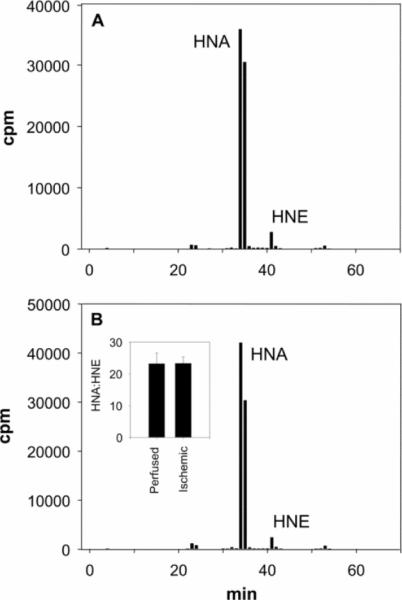
Myocardial ischaemia is associated with the generation of lipid peroxidation products such as HNE (4-hydroxy-trans-2-nonenal); however, the processes that predispose the ischaemic heart to toxicity by HNE and related species are not well understood. In the present study, we examined HNE metabolism in isolated aerobic and ischaemic rat hearts. In aerobic hearts, the reagent [(3)H]HNE was glutathiolated, oxidized to [(3)H]4-hydroxynonenoic acid, and reduced to [(3)H]1,4-dihydroxynonene. In ischaemic hearts, [(3)H]4-hydroxynonenoic acid formation was inhibited and higher levels of [(3)H]1,4-dihydroxynonene and [(3)H]GS-HNE (glutathione conjugate of HNE) were generated. Metabolism of [(3)H]HNE to [(3)H]4-hydroxynonenoic acid was restored upon reperfusion. Reperfused hearts were more efficient at metabolizing HNE than non-ischaemic hearts. Ischaemia increased the myocardial levels of endogenous HNE and 1,4-dihydroxynonene, but not 4-hydroxynonenoic acid. Isolated cardiac mitochondria metabolized [(3)H]HNE primarily to [(3)H]4-hydroxynonenoic acid and minimally to [(3)H]1,4-dihydroxynonene and [(3)H]GS-HNE. Moreover, [(3)H]4-hydroxynonenoic acid was extruded from mitochondria, whereas other [(3)H]HNE metabolites were retained in the matrix. Mitochondria isolated from ischaemic hearts were found to contain 2-fold higher levels of protein-bound HNE than the cytosol, as well as increased [(3)H]GS-HNE and [(3)H]1,4-dihydroxynonene, but not [(3)H]4-hydroxynonenoic acid. Mitochondrial HNE oxidation was inhibited at an NAD(+)/NADH ratio of 0.4 (equivalent to the ischaemic heart) and restored at an NAD(+)/NADH ratio of 8.6 (equivalent to the reperfused heart). These results suggest that HNE metabolism is inhibited during myocardial ischaemia owing to NAD(+) depletion. This decrease in mitochondrial metabolism of lipid peroxidation products and the inability of the mitochondria to extrude HNE metabolites could contribute to myocardial ischaemia/reperfusion injury.
Figures
Similar articles
-
Metabolism of 4-hydroxy-trans-2-nonenal by central nervous system mitochondria is dependent on age and NAD+ availability.Chem Res Toxicol. 2004 Sep;17(9):1272-9. doi: 10.1021/tx049843k. Chem Res Toxicol. 2004. PMID: 15377162
-
Identification of metabolic pathways of the lipid peroxidation product 4-hydroxynonenal by mitochondria isolated from rat kidney cortex.FEBS Lett. 1994 Sep 19;352(1):84-6. doi: 10.1016/0014-5793(94)00922-8. FEBS Lett. 1994. PMID: 7925950
-
Peroxisomal fatty acid oxidation capacity is more resistant to ischaemic and reperfusion injury than mitochondrial fatty acid oxidation capacity in feline hearts.Acta Physiol Scand. 1996 Jun;157(2):133-45. doi: 10.1046/j.1365-201X.1996.498243000.x. Acta Physiol Scand. 1996. PMID: 8800353
-
Intracellular metabolism of 4-hydroxynonenal.Mol Aspects Med. 2003 Aug-Oct;24(4-5):167-75. doi: 10.1016/s0098-2997(03)00011-6. Mol Aspects Med. 2003. PMID: 12892994 Review.
-
Metabolic disarrangement in ischemic heart disease and its therapeutic control.Rev Port Cardiol. 1998 Sep;17(9):667-84. Rev Port Cardiol. 1998. PMID: 9834638 Review.
Cited by
-
A Synthetic Epoxydocosapentaenoic Acid Analogue Ameliorates Cardiac Ischemia/Reperfusion Injury: The Involvement of the Sirtuin 3-NLRP3 Pathway.Int J Mol Sci. 2020 Jul 24;21(15):5261. doi: 10.3390/ijms21155261. Int J Mol Sci. 2020. PMID: 32722183 Free PMC article.
-
Mitochondria as a Source and a Target for Uremic Toxins.Int J Mol Sci. 2019 Jun 25;20(12):3094. doi: 10.3390/ijms20123094. Int J Mol Sci. 2019. PMID: 31242575 Free PMC article. Review.
-
Redox proteomic identification of HNE-bound mitochondrial proteins in cardiac tissues reveals a systemic effect on energy metabolism after doxorubicin treatment.Free Radic Biol Med. 2014 Jul;72:55-65. doi: 10.1016/j.freeradbiomed.2014.03.001. Epub 2014 Mar 12. Free Radic Biol Med. 2014. PMID: 24632380 Free PMC article.
-
Activation of aldehyde dehydrogenase 2 (ALDH2) confers cardioprotection in protein kinase C epsilon (PKCvarepsilon) knockout mice.J Mol Cell Cardiol. 2010 Apr;48(4):757-64. doi: 10.1016/j.yjmcc.2009.10.030. Epub 2009 Nov 11. J Mol Cell Cardiol. 2010. PMID: 19913552 Free PMC article.
-
Protein S-glutathiolation: redox-sensitive regulation of protein function.J Mol Cell Cardiol. 2012 Mar;52(3):559-67. doi: 10.1016/j.yjmcc.2011.07.009. Epub 2011 Jul 20. J Mol Cell Cardiol. 2012. PMID: 21784079 Free PMC article. Review.
References
-
- Downey JM. Free radicals and their involvement during long-term myocardial ischemia and reperfusion. Annu. Rev. Physiol. 1990;52:487–504. - PubMed
-
- Kloner RA, Jennings RB. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 2. Circulation. 2001;104:3158–3167. - PubMed
-
- Chen Z, Siu B, Ho YS, Vincent R, Chua CC, Hamdy RC, Chua BH. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J. Mol. Cell. Cardiol. 1998;30:2281–2289. - PubMed
-
- Li G, Chen Y, Saari JT, Kang YJ. Catalase-overexpressing transgenic mouse heart is resistant to ischemia-reperfusion injury. Am. J. Physiol. 1997;273:H1090–H1095. - PubMed
-
- Chen EP, Bittner HB, Davis RD, Folz RJ, Van Trigt P. Extracellular superoxide dismutase transgene overexpression preserves postischemic myocardial function in isolated murine hearts. Circulation. 1996;94:II412–II417. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources