

A probabilistic framework to predict protein function from interaction data integrated with semantic knowledge
- PMID: 18801191
- PMCID: PMC2570367
- DOI: 10.1186/1471-2105-9-382
A probabilistic framework to predict protein function from interaction data integrated with semantic knowledge
Abstract
Background: The functional characterization of newly discovered proteins has been a challenge in the post-genomic era. Protein-protein interactions provide insights into the functional analysis because the function of unknown proteins can be postulated on the basis of their interaction evidence with known proteins. The protein-protein interaction data sets have been enriched by high-throughput experimental methods. However, the functional analysis using the interaction data has a limitation in accuracy because of the presence of the false positive data experimentally generated and the interactions that are a lack of functional linkage.
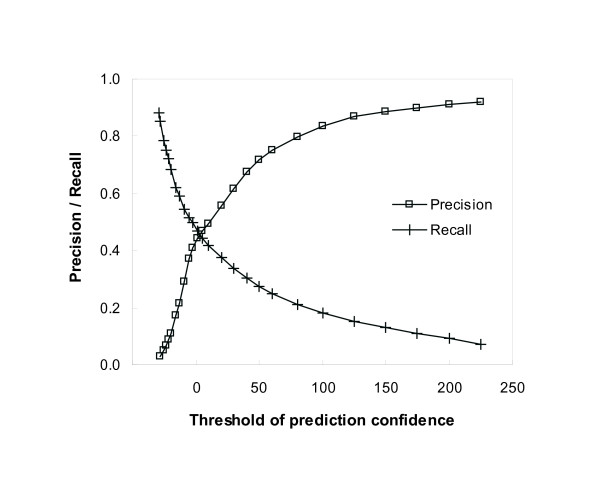
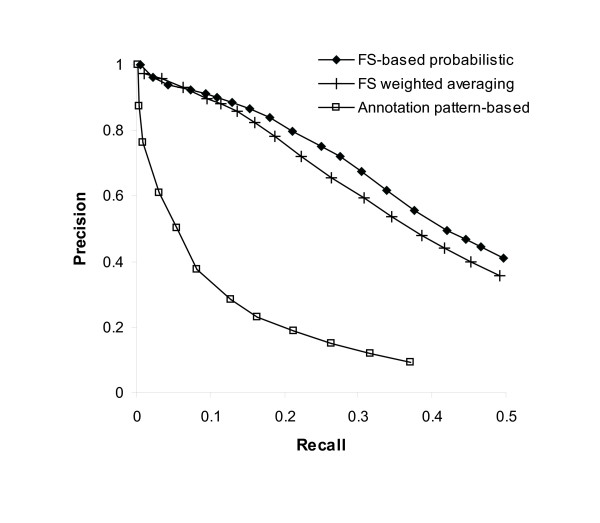
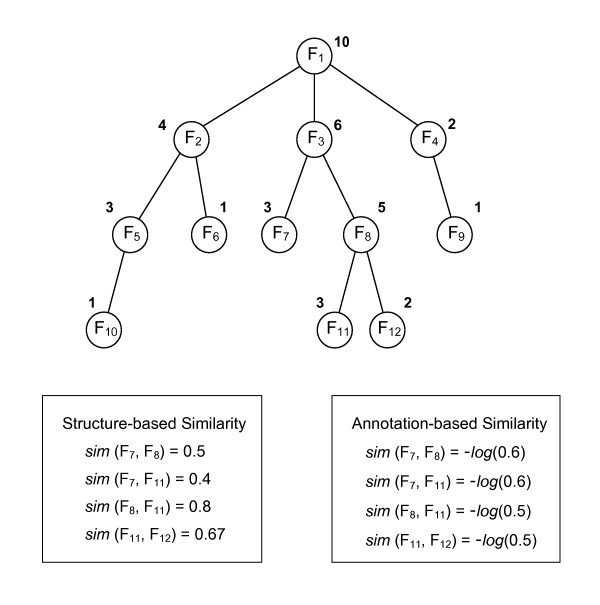
Results: Protein-protein interaction data can be integrated with the functional knowledge existing in the Gene Ontology (GO) database. We apply similarity measures to assess the functional similarity between interacting proteins. We present a probabilistic framework for predicting functions of unknown proteins based on the functional similarity. We use the leave-one-out cross validation to compare the performance. The experimental results demonstrate that our algorithm performs better than other competing methods in terms of prediction accuracy. In particular, it handles the high false positive rates of current interaction data well.
Conclusion: The experimentally determined protein-protein interactions are erroneous to uncover the functional associations among proteins. The performance of function prediction for uncharacterized proteins can be enhanced by the integration of multiple data sources available.
Figures
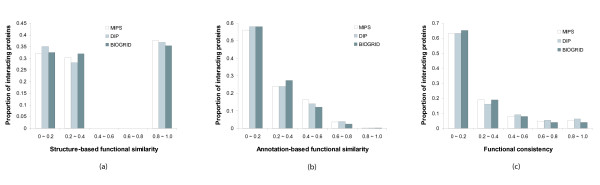
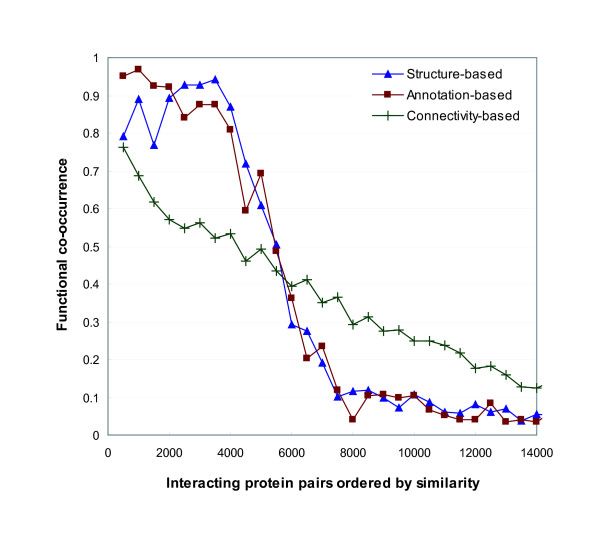
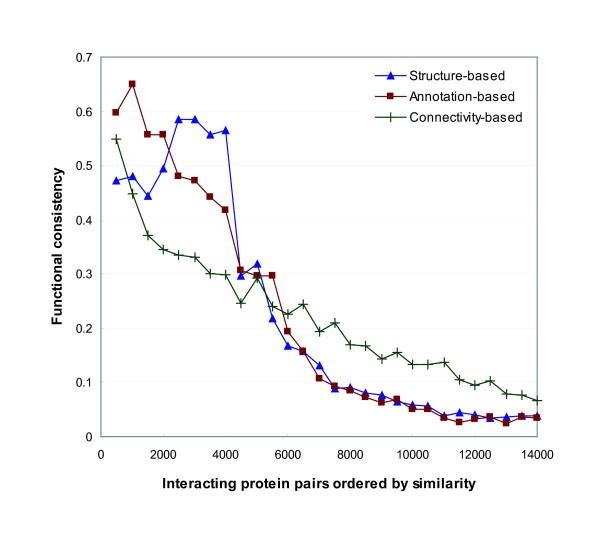
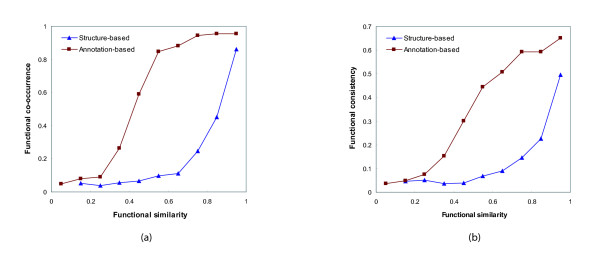
References
-
- Altschul SF, Gish W, Miller W, Meyers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
