

Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation
- PMID: 18802025
- PMCID: PMC2661871
- DOI: 10.1161/CIRCRESAHA.108.178970
Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation
Abstract
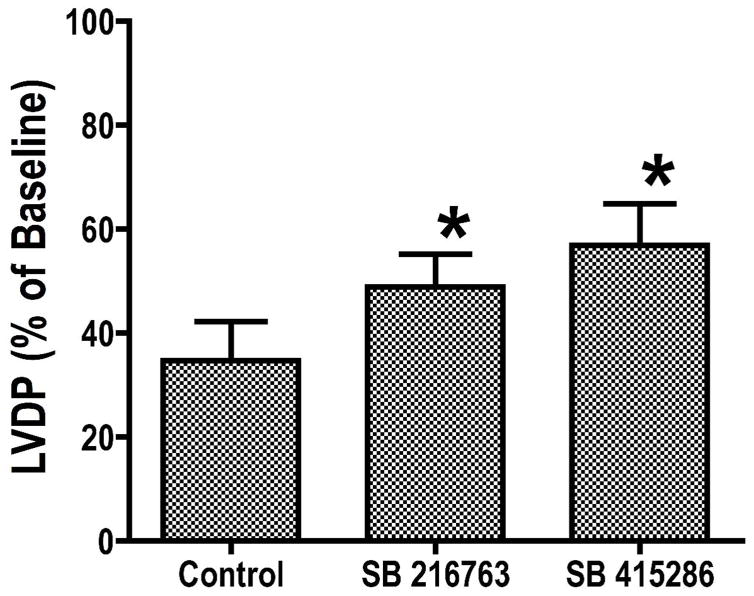
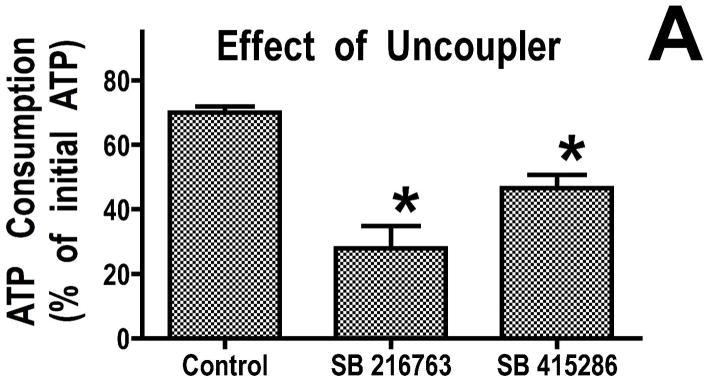
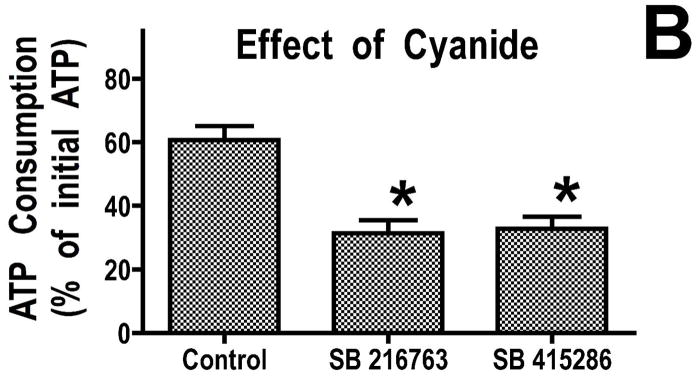
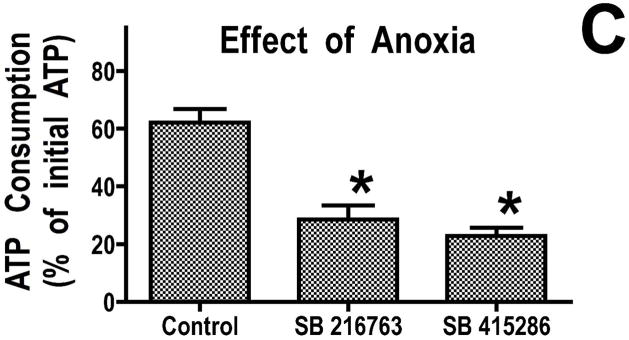
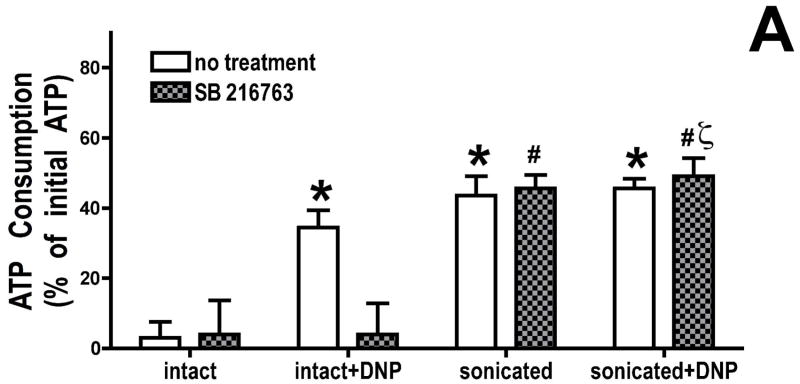
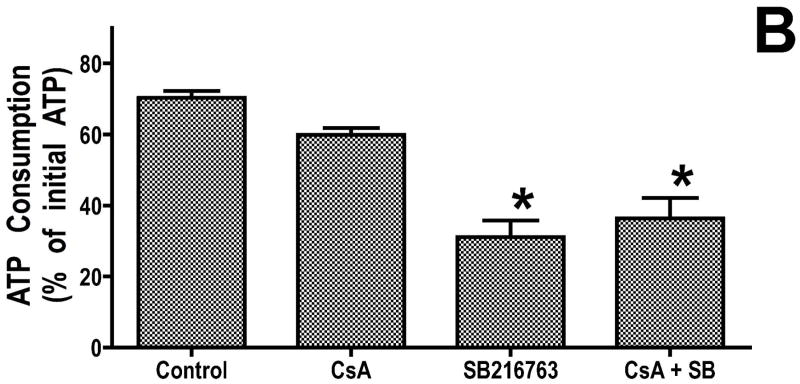
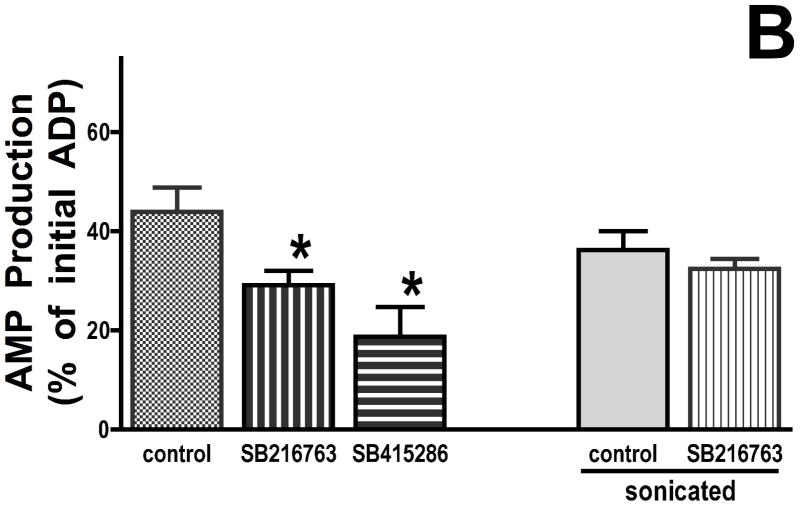
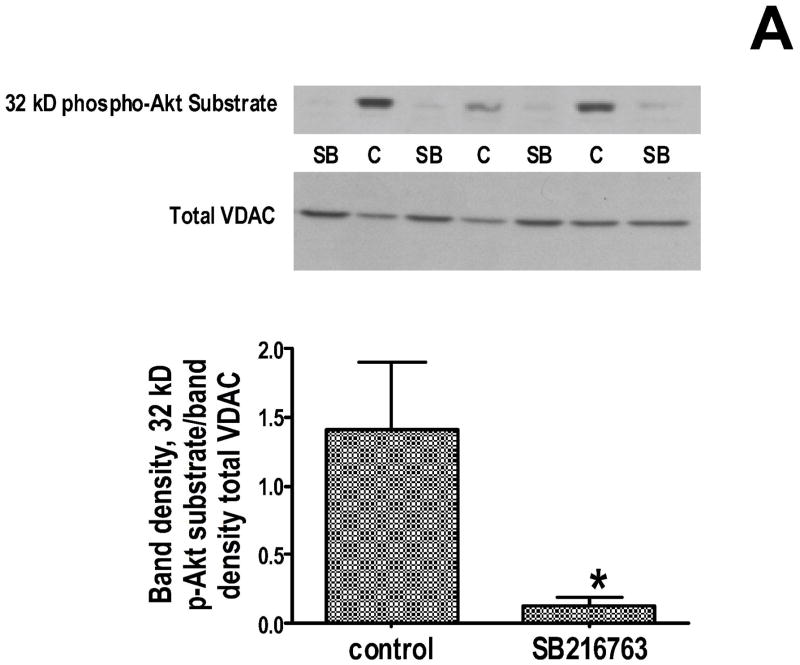
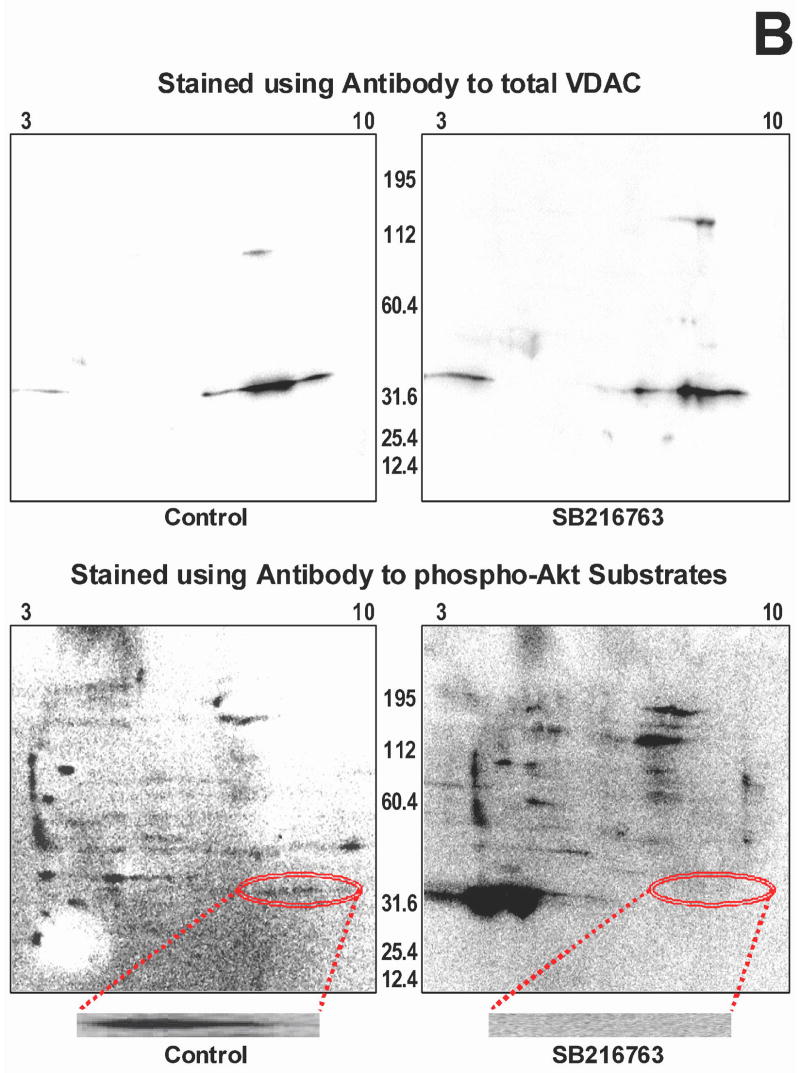
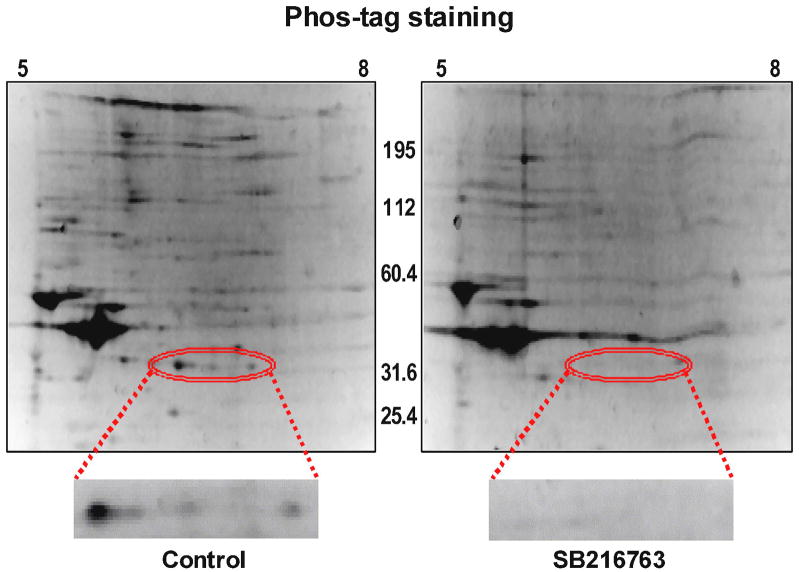
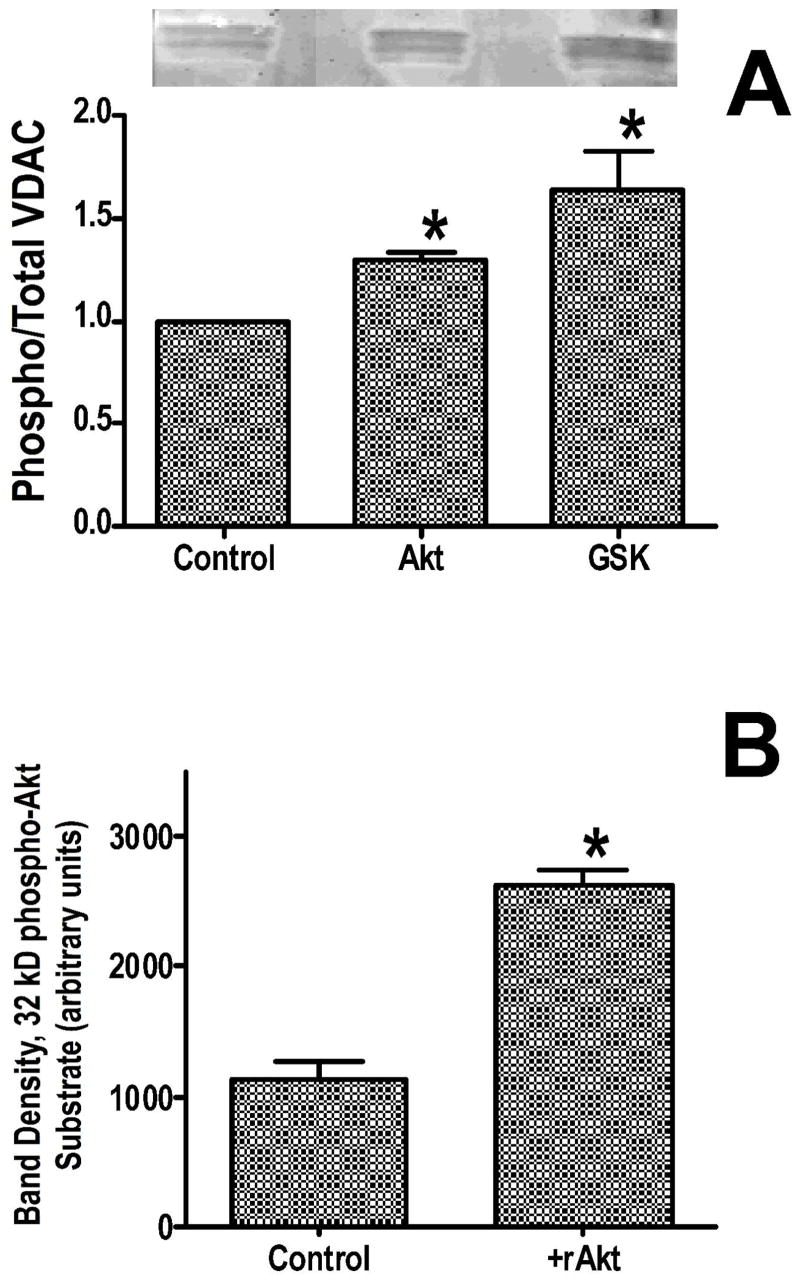
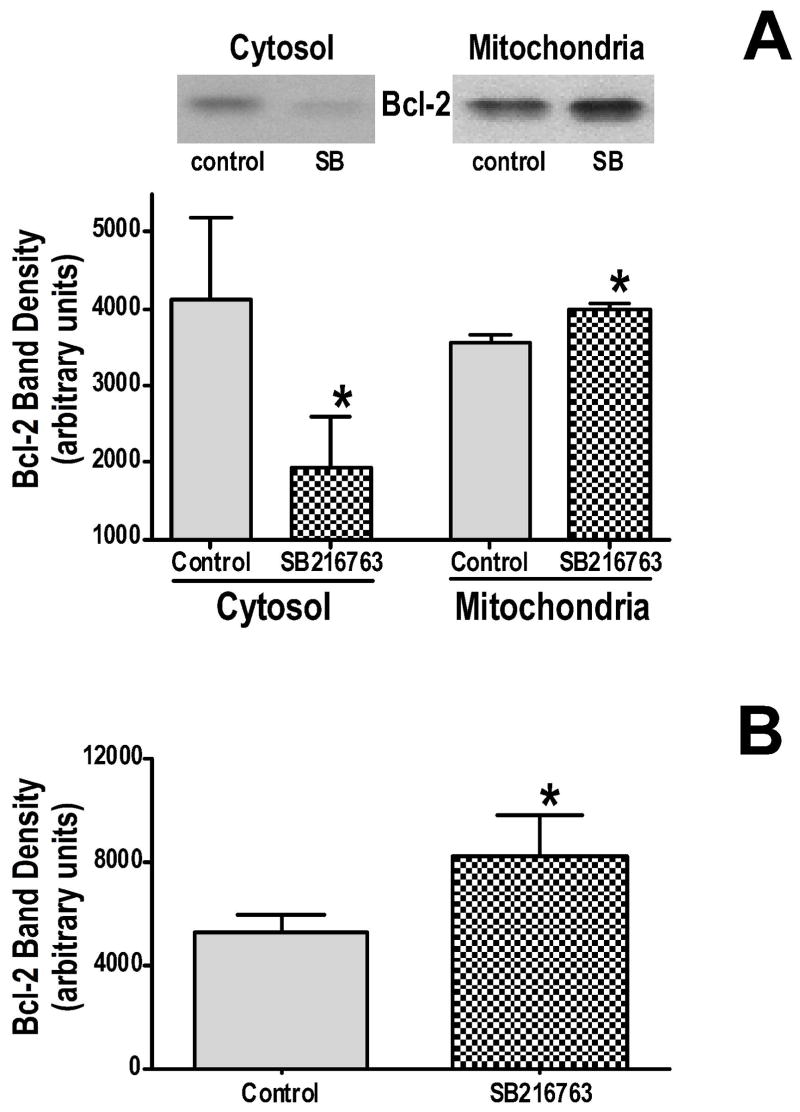
Inhibition of glycogen synthase kinase (GSK)-3 reduces ischemia/reperfusion injury by mechanisms that involve the mitochondria. The goal of this study was to explore possible molecular targets and mechanistic basis of this cardioprotective effect. In perfused rat hearts, treatment with GSK inhibitors before ischemia significantly improved recovery of function. To assess the effect of GSK inhibitors on mitochondrial function under ischemic conditions, mitochondria were isolated from rat hearts perfused with GSK inhibitors and were treated with uncoupler or cyanide or were made anoxic. GSK inhibition slowed ATP consumption under these conditions, which could be attributable to inhibition of ATP entry into the mitochondria through the voltage-dependent anion channel (VDAC) and/or adenine nucleotide transporter (ANT) or to inhibition of the F(1)F(0)-ATPase. To determine the site of the inhibitory effect on ATP consumption, we measured the conversion of ADP to AMP by adenylate kinase located in the intermembrane space. This assay requires adenine nucleotide transport across the outer but not the inner mitochondrial membrane, and we found that GSK inhibitors slow AMP production similar to their effect on ATP consumption. This suggests that GSK inhibitors are acting on outer mitochondrial membrane transport. In sonicated mitochondria, GSK inhibition had no effect on ATP consumption or AMP production. In intact mitochondria, cyclosporin A had no effect, indicating that ATP consumption is not caused by opening of the mitochondrial permeability transition pore. Because GSK is a kinase, we assessed whether protein phosphorylation might be involved. Therefore, we performed Western blot and 1D/2D gel phosphorylation site analysis using phos-tag staining to indicate proteins that had decreased phosphorylation in hearts treated with GSK inhibitors. Liquid chromatographic-mass spectrometric analysis revealed 1 of these proteins to be VDAC2. Taken together, we found that GSK-mediated signaling modulates transport through the outer membrane of the mitochondria. Both proteomics and adenine nucleotide transport data suggest that GSK regulates VDAC and that VDAC may be an important regulatory site in ischemia/reperfusion injury.
Conflict of interest statement
Figures

Comment in
-
Overcoming an energy crisis?: an adaptive role of glycogen synthase kinase-3 inhibition in ischemia/reperfusion.Circ Res. 2008 Oct 24;103(9):910-3. doi: 10.1161/01.RES.0000338259.37472.b6. Circ Res. 2008. PMID: 18948628 Free PMC article. No abstract available.
References
-
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. - PubMed
-
- Gross ER, Gross GJ. Ligand triggers of classical preconditioning and postconditioning. Cardiovasc Res. 2006;70:212–221. - PubMed
-
- Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev. 2007;12:181–188. - PubMed
-
- Tong H, Rockman HA, Koch WJ, Steenbergen C, Murphy E. G protein-coupled receptor internalization signaling is required for cardioprotection in ischemic preconditioning. Circ Res. 2004;94:1133–1141. - PubMed
-
- Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signaling: taking a RISK for cardioprotection. Heart Fail Rev. 2007;12:217–234. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
