

Neutrophil activation by the tissue factor/Factor VIIa/PAR2 axis mediates fetal death in a mouse model of antiphospholipid syndrome
- PMID: 18802482
- PMCID: PMC2542852
- DOI: 10.1172/JCI36089
Neutrophil activation by the tissue factor/Factor VIIa/PAR2 axis mediates fetal death in a mouse model of antiphospholipid syndrome
Abstract
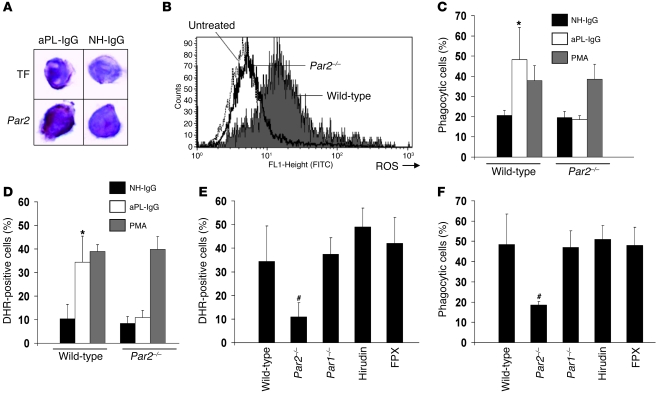
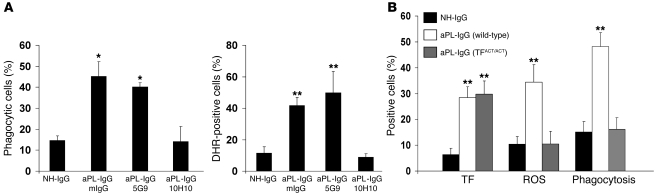
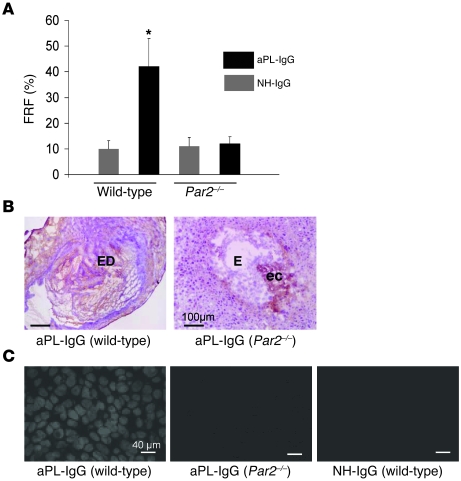
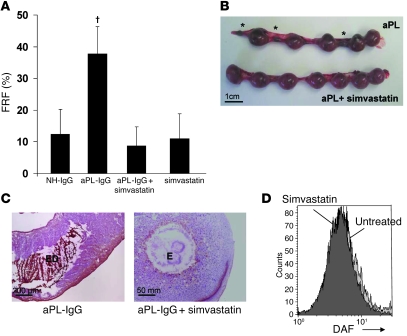
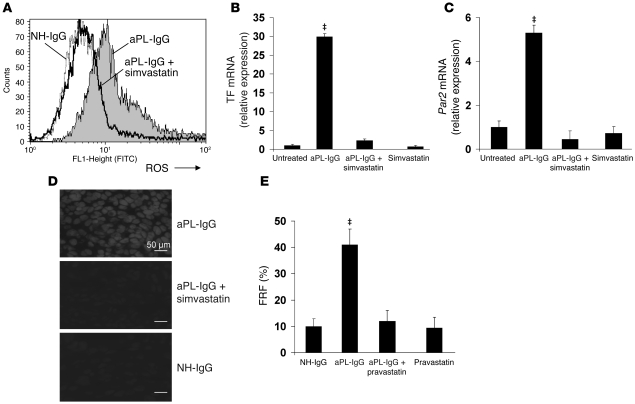
Women with antiphospholipid syndrome (APS), a condition characterized by the presence of antiphospholipid antibodies (aPL), often suffer pregnancy-related complications, including miscarriage. We have previously shown that C5a induction of tissue factor (TF) expression in neutrophils contributes to respiratory burst, trophoblast injury, and pregnancy loss in mice treated with aPL. Here we analyzed how TF contributes to neutrophil activation and trophoblast injury in this model. Neutrophils from aPL-treated mice expressed protease-activated receptor 2 (PAR2), and stimulation of this receptor led to neutrophil activation, trophoblast injury, and fetal death. An antibody specific for human TF that has little impact on coagulation, but potently inhibits TF/Factor VIIa (FVIIa) signaling through PAR2, inhibited aPL-induced neutrophil activation in mice that expressed human TF. Genetic deletion of the TF cytoplasmic domain, which allows interaction between TF and PAR2, reduced aPL-induced neutrophil activation in aPL-treated mice. Par2-/- mice treated with aPL exhibited reduced neutrophil activation and normal pregnancies, which indicates that PAR2 plays an important role in the pathogenesis of aPL-induced fetal injury. We also demonstrated that simvastatin and pravastatin decreased TF and PAR2 expression on neutrophils and prevented pregnancy loss. Our results suggest that TF/FVIIa/PAR2 signaling mediates neutrophil activation and fetal death in APS and that statins may be a good treatment for women with aPL-induced pregnancy complications.
Figures
Comment in
-
Tracing the molecular pathogenesis of antiphospholipid syndrome.J Clin Invest. 2008 Oct;118(10):3276-8. doi: 10.1172/JCI37243. J Clin Invest. 2008. PMID: 18802489 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
