

How does the Schwann cell lineage form tumors in NF1?
- PMID: 18803326
- PMCID: PMC2652636
- DOI: 10.1002/glia.20776
How does the Schwann cell lineage form tumors in NF1?
Abstract
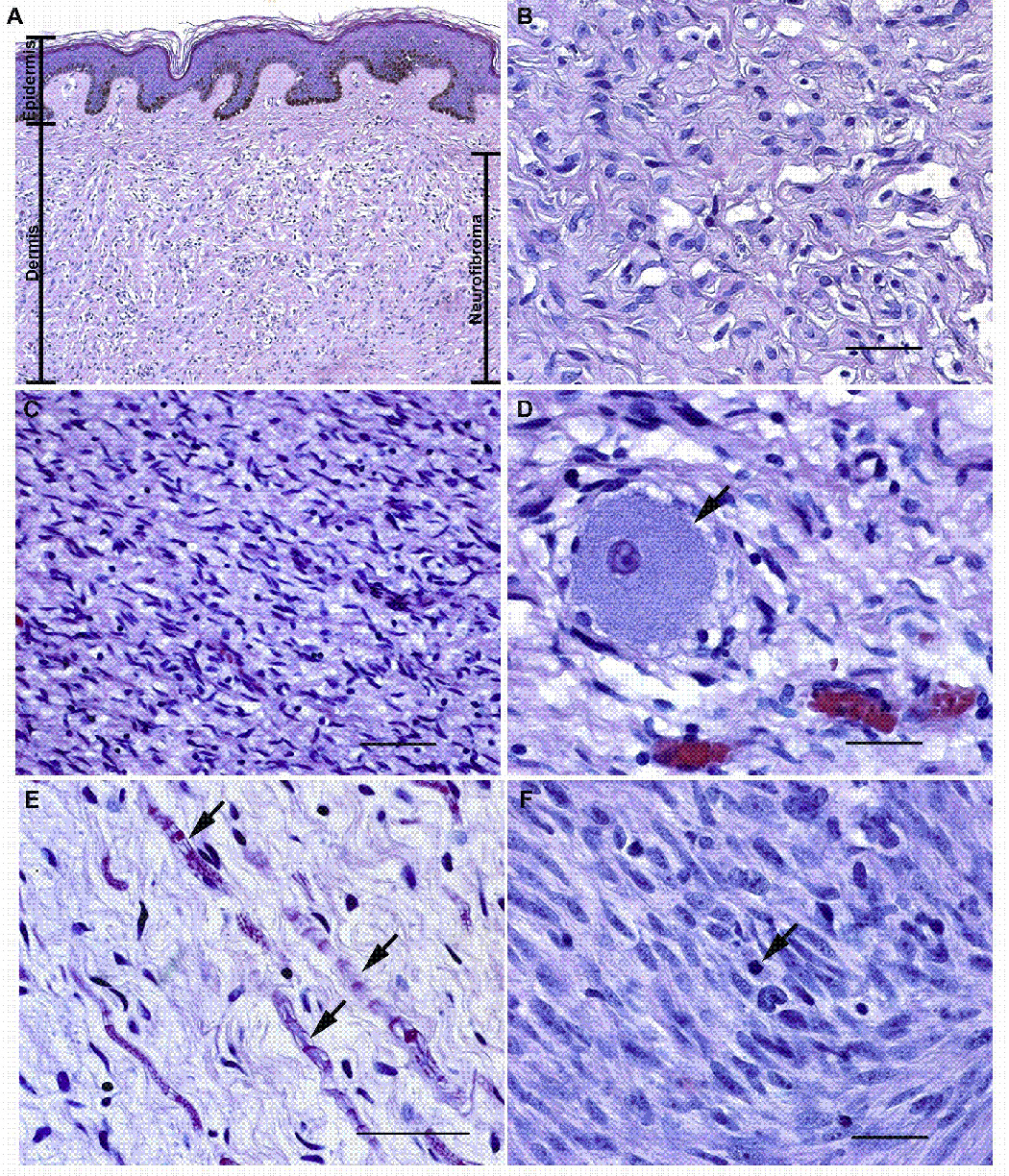
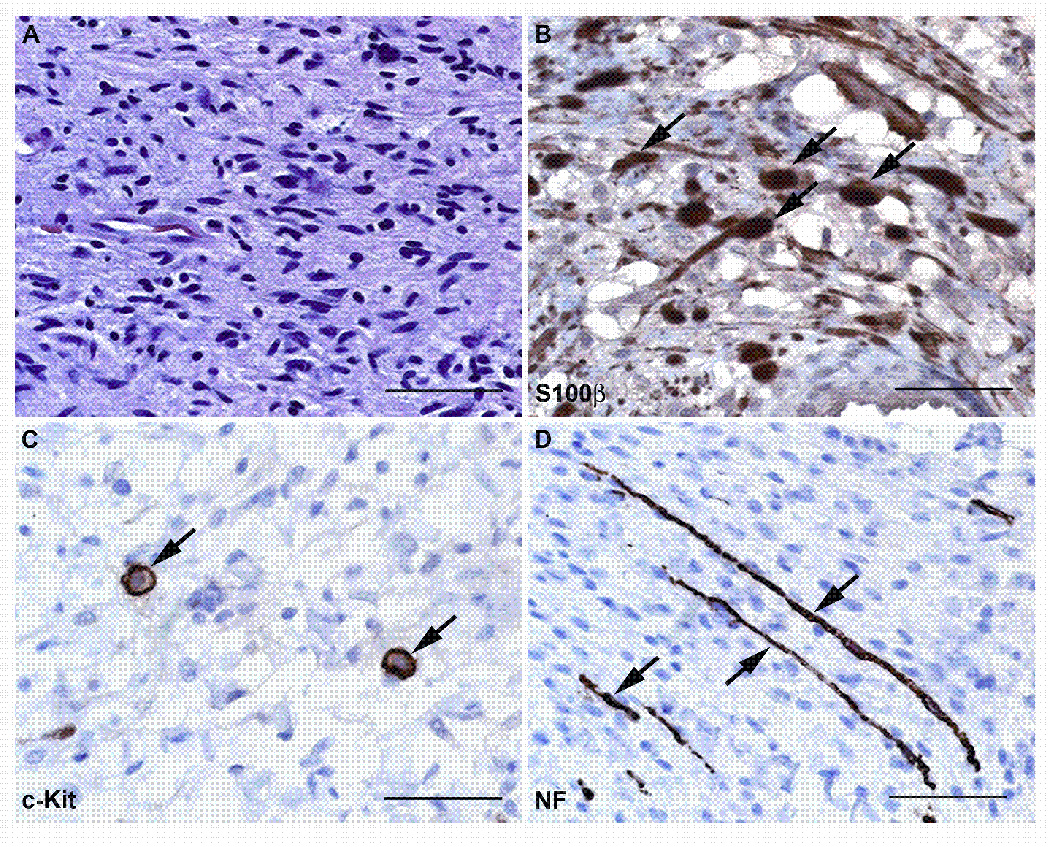
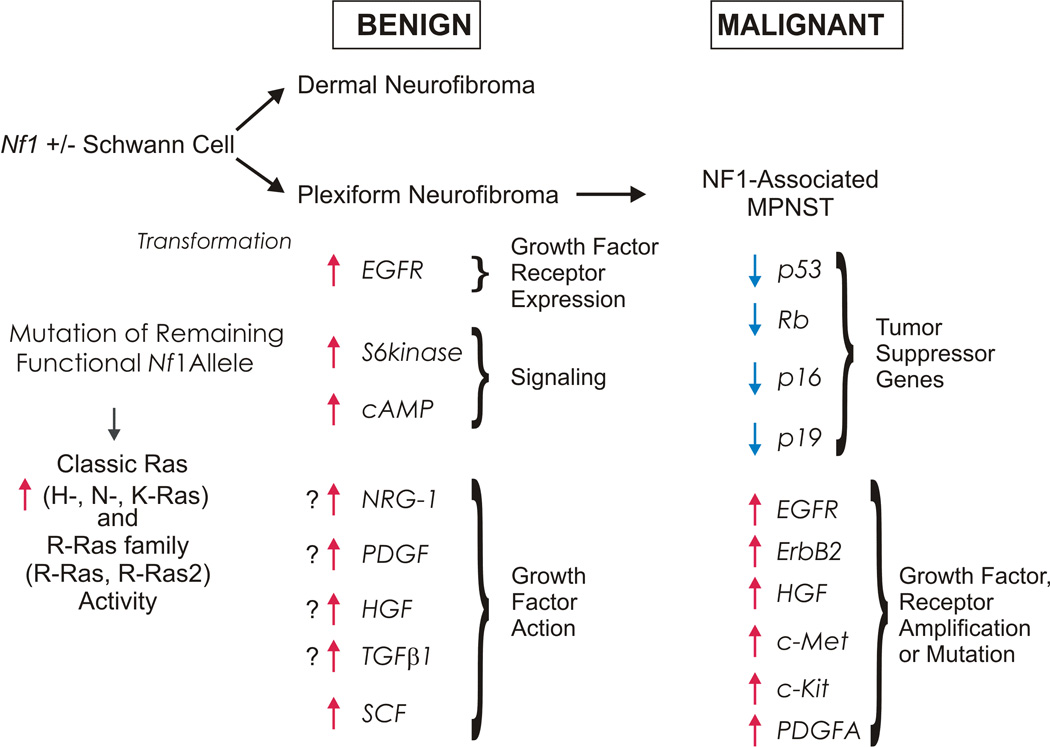
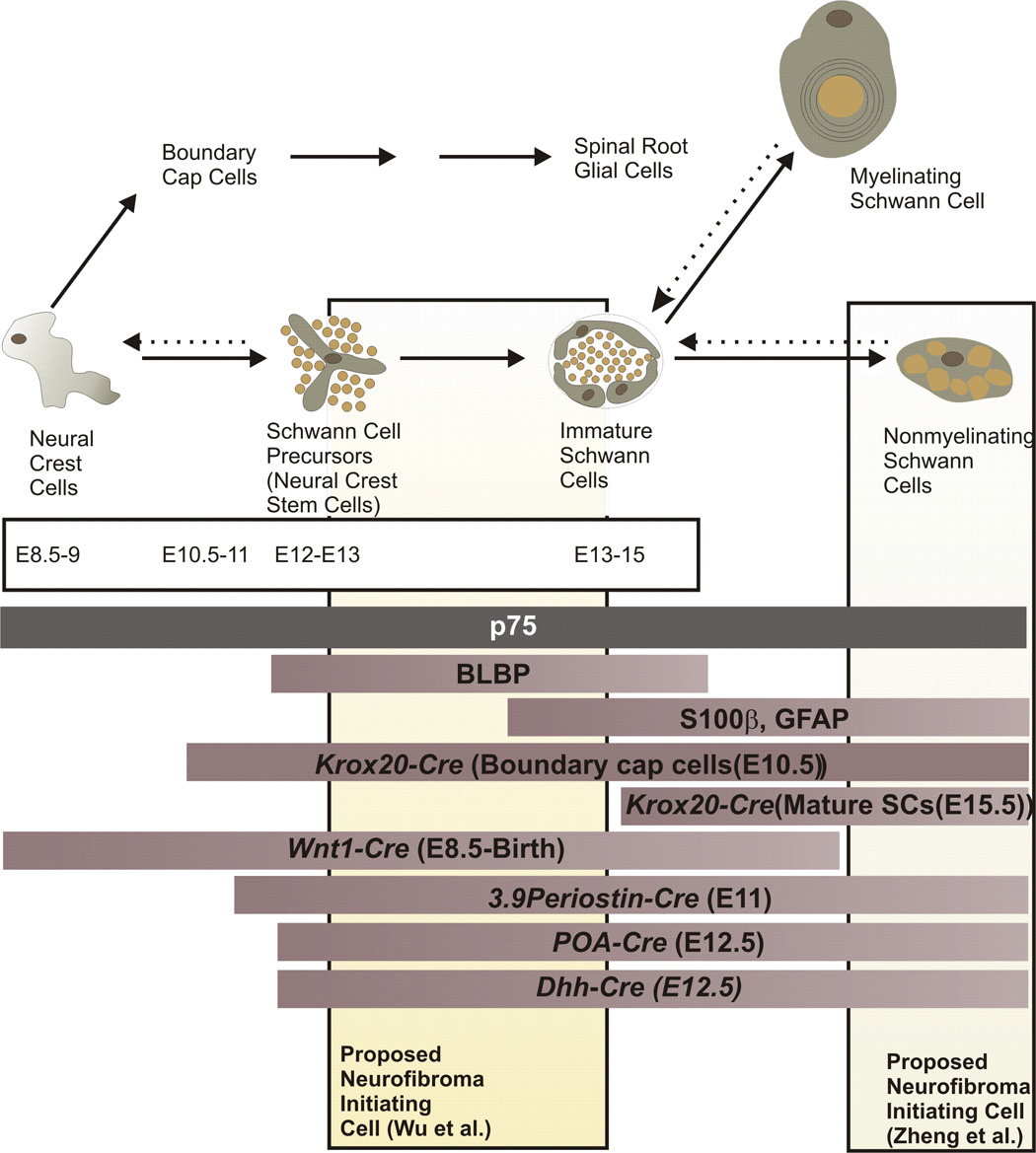
Neurofibromas are benign tumors of peripheral nerve that occur sporadically or in patients with the autosomal dominant tumor predisposition syndrome neurofibromatosis type 1 (NF1). Multiple neurofibroma subtypes exist which differ in their site of occurrence, their association with NF1, and their tendency to undergo transformation to become malignant peripheral nerve sheath tumors (MPNSTs), the most common malignancy associated with NF1. Most NF1 patients carry a constitutional mutation of the NF1 tumor suppressor gene. Neurofibromas develop in these patients when an unknown cell type in the Schwann cell lineage loses its remaining functional NF1 gene and initiates a complex series of interactions with other cell types; these interactions may be influenced by aberrant expression of growth factors and growth factor receptors and the action of modifier genes. Cells within certain neurofibroma subtypes subsequently accumulate additional mutations affecting the p19(ARF)-MDM2-TP53 and p16INK4A-Rb signaling cascades, mutations of other as yet unidentified genes, and amplification of growth factor receptor genes, resulting in their transformation into MPNSTs. These observations have been validated using a variety of transgenic and knockout mouse models that recapitulate neurofibroma and MPNST pathogenesis. A new generation of mouse models is also providing important new insights into the identity of the cell type in the Schwann cell lineage that gives rise to neurofibromas. Our improving understanding of the mechanisms underlying the pathogenesis of neurofibromas and MPNSTs raises intriguing new questions about the origin and pathogenesis of these neoplasms and establishes models for the development of new therapies targeting these neoplasms.
Figures
References
-
- Aoki M, Nabeshima K, Koga K, Hamasaki M, Suzumiya J, Tamura K, Iwasaki H. Imatinib mesylate inhibits cell invasion of malignant peripheral nerve sheath tumor induced by platelet-derived growth factor-BB. Lab Invest. 2007;87:767–779. - PubMed
-
- Aquino J, Hjerling-Leffler J, Koltzenburg M, Villar M, Ernfors P. In vitro and in vivo differentiation of boundary cap neural crest stem cells into mature Schwann cells. Exp Neurol. 2006;198:438–449. - PubMed
-
- Badache A, DeVries GH. Neurofibrosarcoma-derived Schwann cells overexpress platelet-derived growth factor (PDGF) receptors and are induced to proliferate by PDGF BB. J Cell Physiol. 1998;177:334–342. - PubMed
-
- Badache A, Muja N, DeVries GH. Expression of Kit in neurofibromin-deficient human Schwann cells: role in Schwann cell hyperplasia associated with type 1 neurofibromatosis. Oncogene. 1998;17:795–800. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
