

The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes
- PMID: 18803844
- PMCID: PMC2563014
- DOI: 10.1186/1471-2105-9-386
The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes
Abstract
Background: Random community genomes (metagenomes) are now commonly used to study microbes in different environments. Over the past few years, the major challenge associated with metagenomics shifted from generating to analyzing sequences. High-throughput, low-cost next-generation sequencing has provided access to metagenomics to a wide range of researchers.
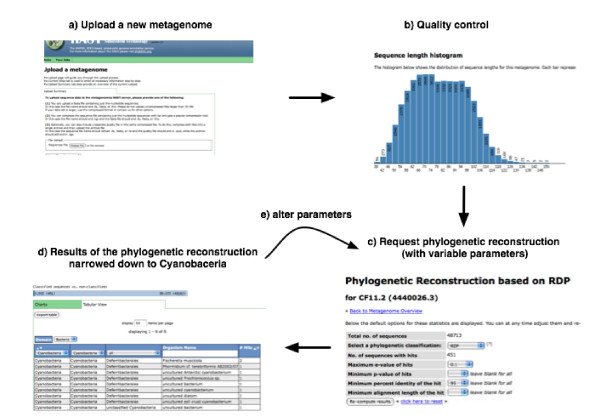
Results: A high-throughput pipeline has been constructed to provide high-performance computing to all researchers interested in using metagenomics. The pipeline produces automated functional assignments of sequences in the metagenome by comparing both protein and nucleotide databases. Phylogenetic and functional summaries of the metagenomes are generated, and tools for comparative metagenomics are incorporated into the standard views. User access is controlled to ensure data privacy, but the collaborative environment underpinning the service provides a framework for sharing datasets between multiple users. In the metagenomics RAST, all users retain full control of their data, and everything is available for download in a variety of formats.
Conclusion: The open-source metagenomics RAST service provides a new paradigm for the annotation and analysis of metagenomes. With built-in support for multiple data sources and a back end that houses abstract data types, the metagenomics RAST is stable, extensible, and freely available to all researchers. This service has removed one of the primary bottlenecks in metagenome sequence analysis - the availability of high-performance computing for annotating the data. http://metagenomics.nmpdr.org.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
