

The removal of multiplicative, systematic bias allows integration of breast cancer gene expression datasets - improving meta-analysis and prediction of prognosis
- PMID: 18803878
- PMCID: PMC2563019
- DOI: 10.1186/1755-8794-1-42
The removal of multiplicative, systematic bias allows integration of breast cancer gene expression datasets - improving meta-analysis and prediction of prognosis
Abstract
Background: The number of gene expression studies in the public domain is rapidly increasing, representing a highly valuable resource. However, dataset-specific bias precludes meta-analysis at the raw transcript level, even when the RNA is from comparable sources and has been processed on the same microarray platform using similar protocols. Here, we demonstrate, using Affymetrix data, that much of this bias can be removed, allowing multiple datasets to be legitimately combined for meaningful meta-analyses.
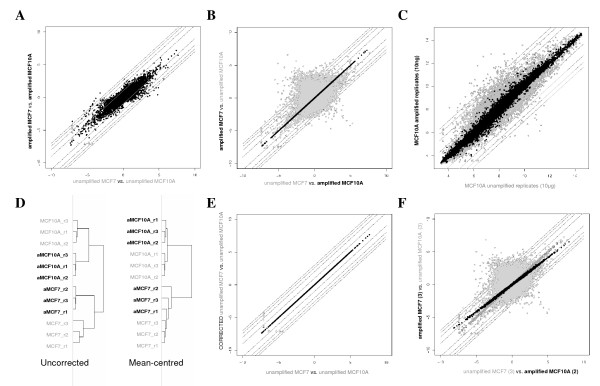
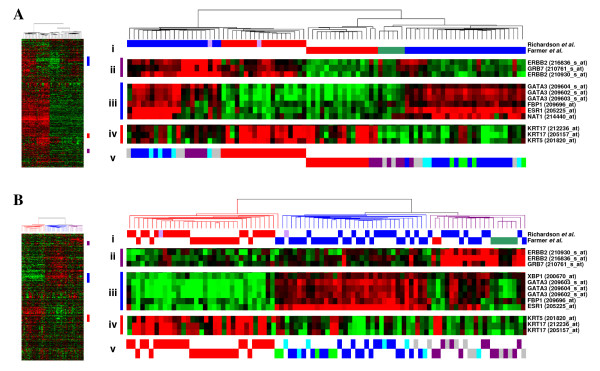
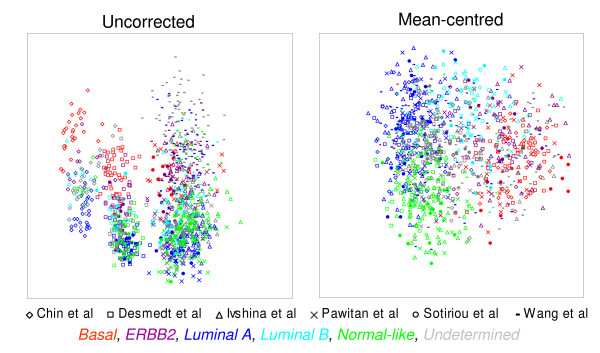
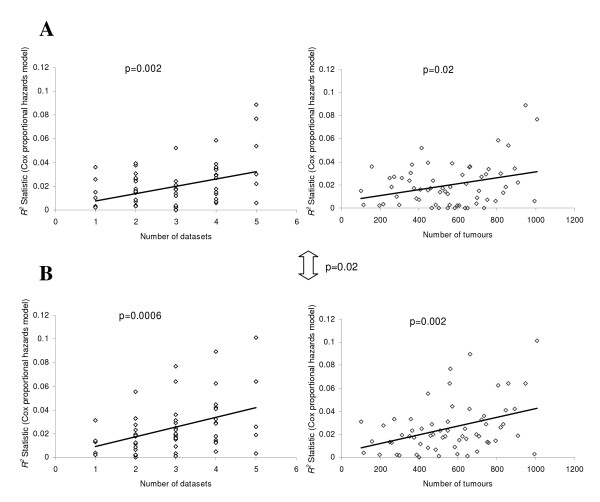
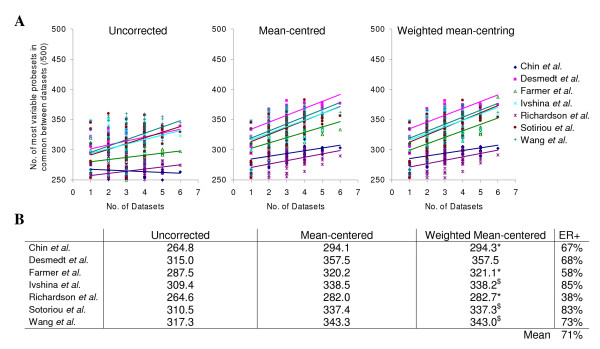
Results: A series of validation datasets comparing breast cancer and normal breast cell lines (MCF7 and MCF10A) were generated to examine the variability between datasets generated using different amounts of starting RNA, alternative protocols, different generations of Affymetrix GeneChip or scanning hardware. We demonstrate that systematic, multiplicative biases are introduced at the RNA, hybridization and image-capture stages of a microarray experiment. Simple batch mean-centering was found to significantly reduce the level of inter-experimental variation, allowing raw transcript levels to be compared across datasets with confidence. By accounting for dataset-specific bias, we were able to assemble the largest gene expression dataset of primary breast tumours to-date (1107), from six previously published studies. Using this meta-dataset, we demonstrate that combining greater numbers of datasets or tumours leads to a greater overlap in differentially expressed genes and more accurate prognostic predictions. However, this is highly dependent upon the composition of the datasets and patient characteristics.
Conclusion: Multiplicative, systematic biases are introduced at many stages of microarray experiments. When these are reconciled, raw data can be directly integrated from different gene expression datasets leading to new biological findings with increased statistical power.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
