

Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration
- PMID: 18804434
- PMCID: PMC2563805
- DOI: 10.1016/j.devcel.2008.06.015
Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration
Abstract
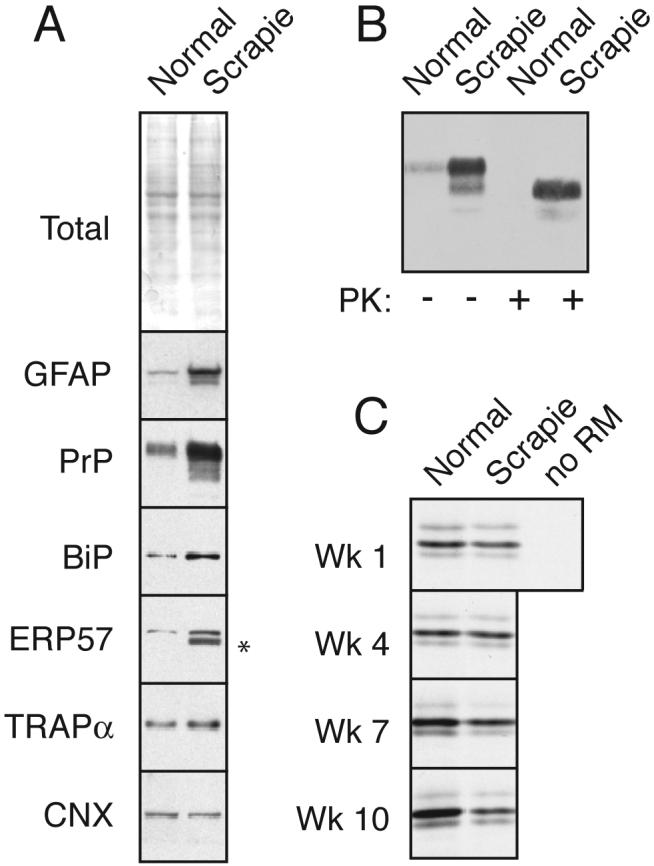
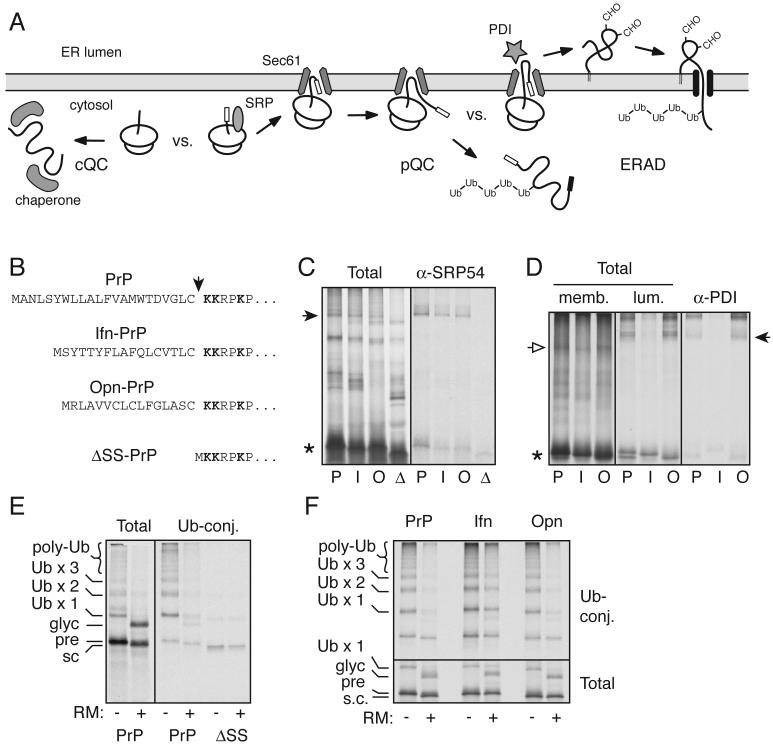
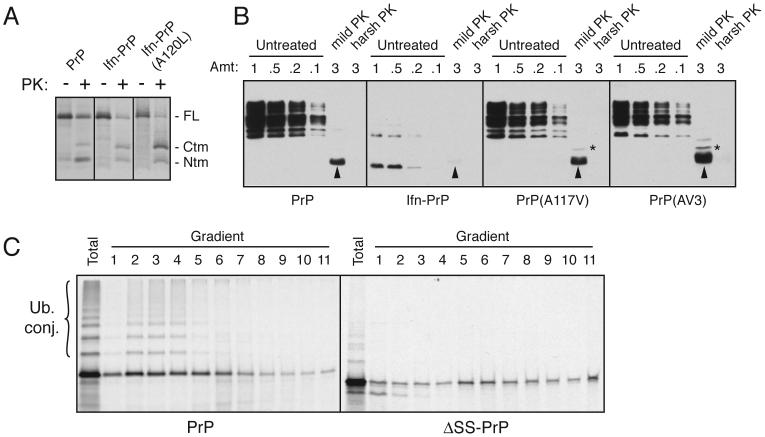
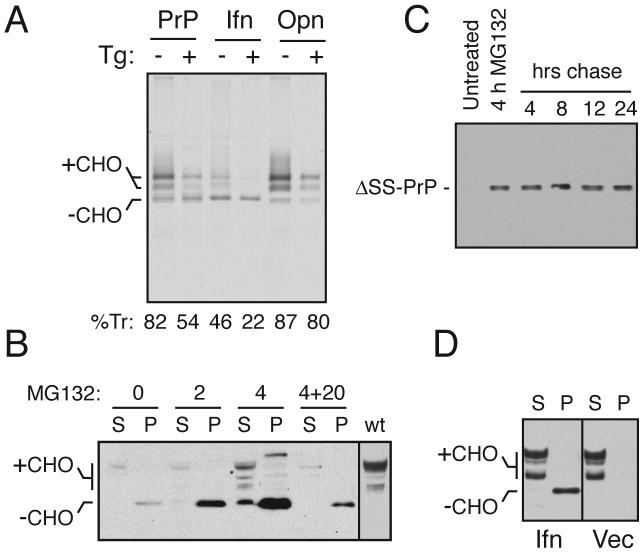
During acute stress in the endoplasmic reticulum (ER), mammalian prion protein (PrP) is temporarily prevented from translocation into the ER and instead routed directly for cytosolic degradation. This "pre-emptive" quality control (pQC) system benefits cells by minimizing PrP aggregation in the secretory pathway during ER stress. However, the potential toxicity of cytosolic PrP raised the possibility that persistent pQC of PrP contributes to neurodegeneration in prion diseases. Here, we find evidence of ER stress and decreased translocation of nascent PrP during prion infection. Transgenic mice expressing a PrP variant with reduced translocation at levels expected during ER stress was sufficient to cause several mild age-dependent clinical and histological manifestations of PrP-mediated neurodegeneration. Thus, an ordinarily adaptive quality-control pathway can be contextually detrimental over long time periods. We propose that one mechanism of prion-mediated neurodegeneration involves an indirect ER stress-dependent effect on nascent PrP biosynthesis and metabolism.
Figures
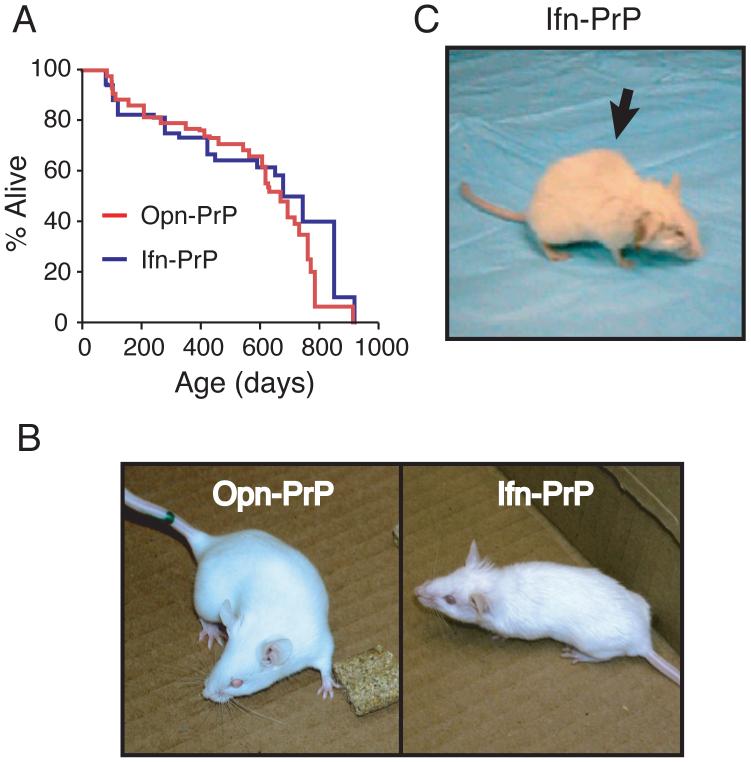
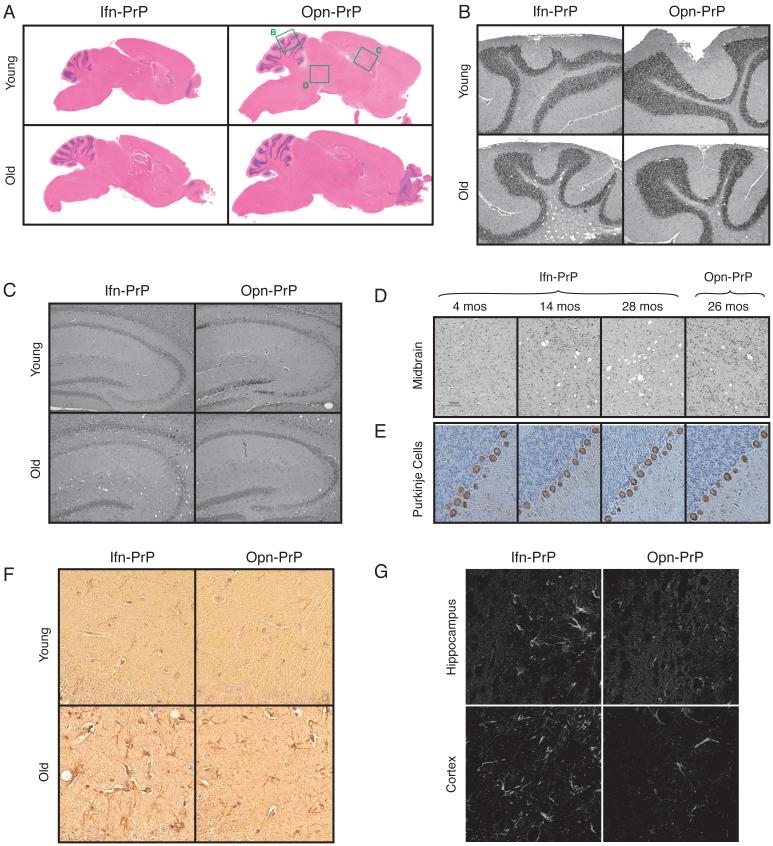
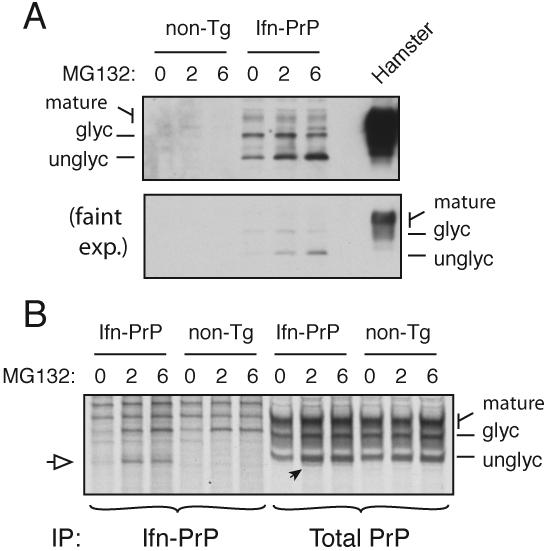
Comment in
-
Endoplasmic reticulum stress, PrP trafficking, and neurodegeneration.Dev Cell. 2008 Sep;15(3):339-341. doi: 10.1016/j.devcel.2008.09.001. Dev Cell. 2008. PMID: 18804431 Free PMC article.
References
-
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. - PubMed
-
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. - PubMed
-
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. - PubMed
-
- Drisaldi B, Stewart RS, Adles C, Stewart LR, Quaglio E, Biasini E, Fioriti L, Chiesa R, Harris DA. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem. 2003;278:21732–21743. - PubMed
-
- Fioriti L, Dossena S, Stewart LR, Stewart RS, Harris DA, Forloni G, Chiesa R. Cytosolic prion protein (PrP) is not toxic in N2a cells and primary neurons expressing pathogenic PrP mutations. J Biol Chem. 2005;280:11320–11328. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
