

Case Reports
doi: 10.1016/j.braindev.2008.08.005.
Epub 2008 Sep 19.
A novel POMT2 mutation causes mild congenital muscular dystrophy with normal brain MRI
Affiliations
- PMID: 18804929
- PMCID: PMC2702532
- DOI: 10.1016/j.braindev.2008.08.005
Item in Clipboard
Case Reports
A novel POMT2 mutation causes mild congenital muscular dystrophy with normal brain MRI
Brain Dev.
2009 Jun.
Abstract
We report a patient harboring a novel homozygous mutation of c.604T>G (p.F202V) in POMT2. He showed delayed psychomotor development but acquired the ability to walk at the age of 3 years and 10 months. His brain MRI was normal. No ocular abnormalities were seen. Biopsied skeletal muscle revealed markedly decreased but still detectable glycosylated forms of alpha-dystroglycan (alpha-DG). Our results indicate that mutations in POMT2 can cause a wide spectrum of clinical phenotypes as observed in other genes associated with alpha-dystroglycanopathy. Presence of small amounts of partly glycosylated alpha-DG may have a role in reducing the clinical symptoms of alpha-dystroglycanopathy.
Figures

(A) The patient can stand and walk with no support. Minimal calf hypertrophy is seen. (B) T2 weighted brain magnetic resonance imaging shows no obvious brain anomaly, cortical dysplasia, or white matter changes. (C) Sequence analysis of POMT2 revealed a homozygous mutation at c.604T>G in exon 5.
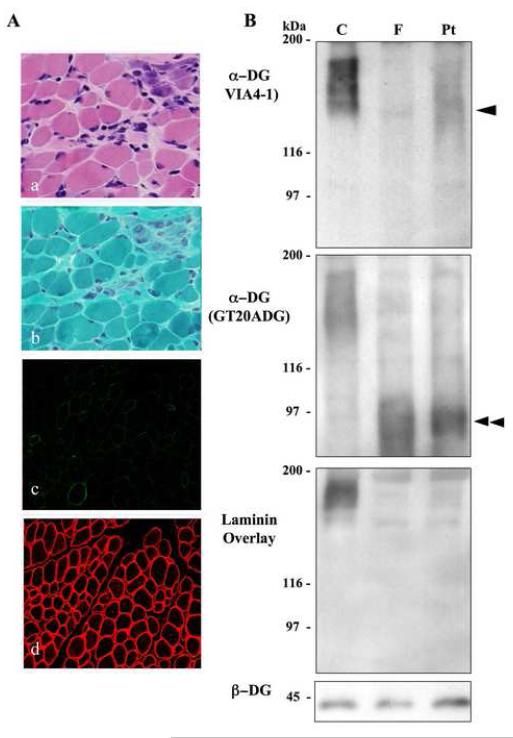
(A) Histological analysis. On Hematoxylin and eosin (a) and modified Gomori-trichrime (b) staining, variation in fiber size and scattered necrotic and regenerating fibers are seen. Immunohistochemical analysis using antibodies VIA4-1 (c), which recognize heavily glycosylated form of α-dystroglycan (α—DG), showed greatly reduced sarcolemmal staining in patient, but well-preserved immunoreactivities of β-DG (d) is seen. Bar = 50m. (B) Immunoblotting analysis. Immunoblotting analysis using antibodies of VIA4-1, GT20ADG for α-dystroglycan (α-DG) and laminin overlay assay are performed using skeletal muscle from control (C), Fukuyama-type congenital muscular dystrophy (FCMD; F), and the patient (Pt). VIA4-1 recognizes a broad band about 156 kDa in size in control, and approximately 90kDa in FCMD. In the patient muscle, reduced in size and amount compared with control was observed. GT20ADG revealed bands at approximately 90 kDa in both the patient and FCMD muscles. Laminin overlay assay shows barely detectable band in both the patient and FCMD.
Similar articles
-
A fourth case of POMT2-related limb girdle muscle dystrophy with mild reduction of α-dystroglycan glycosylation.Eur J Paediatr Neurol. 2014 May;18(3):404-8. doi: 10.1016/j.ejpn.2013.10.005. Epub 2013 Oct 27. Eur J Paediatr Neurol. 2014. PMID: 24183756
-
Congenital muscular dystrophy with muscle inflammation alpha dystroglycan glycosylation defect and no mutation in FKRP gene.J Neurol Sci. 2006 Apr 15;243(1-2):47-51. doi: 10.1016/j.jns.2005.11.024. Epub 2006 Jan 4. J Neurol Sci. 2006. PMID: 16386759
-
Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan.Brain. 2007 Oct;130(Pt 10):2725-35. doi: 10.1093/brain/awm212. Epub 2007 Sep 18. Brain. 2007. PMID: 17878207
-
[Fukuyama congenital muscular dystrophy and related alpha-dystroglycanopathies].Brain Nerve. 2008 Oct;60(10):1159-64. Brain Nerve. 2008. PMID: 18975603 Review. Japanese.
-
Telethonin-deficiency initially presenting as a congenital muscular dystrophy.Neuromuscul Disord. 2011 Jun;21(6):433-8. doi: 10.1016/j.nmd.2011.03.005. Epub 2011 May 6. Neuromuscul Disord. 2011. PMID: 21530252 Review.
Cited by
-
A homozygous mutation in the POMT2 gene in four siblings with limb-girdle muscular dystrophy 2N.Turk Arch Pediatr. 2021 Jan 1;56(1):68-71. doi: 10.14744/TurkPediatriArs.2020.37880. eCollection 2021 Jan. Turk Arch Pediatr. 2021. PMID: 34013233 Free PMC article.
-
Biochemical correlation of activity of the α-dystroglycan-modifying glycosyltransferase POMGnT1 with mutations in muscle-eye-brain disease.Biochem J. 2011 Jun 1;436(2):447-55. doi: 10.1042/BJ20101059. Biochem J. 2011. PMID: 21361872 Free PMC article.
-
Phenotype and Genotype Study of Chinese POMT2-Related α-Dystroglycanopathy.Front Genet. 2021 Aug 3;12:692479. doi: 10.3389/fgene.2021.692479. eCollection 2021. Front Genet. 2021. PMID: 34413876 Free PMC article.
-
Structure of the eukaryotic protein O-mannosyltransferase Pmt1-Pmt2 complex.Nat Struct Mol Biol. 2019 Aug;26(8):704-711. doi: 10.1038/s41594-019-0262-6. Epub 2019 Jul 8. Nat Struct Mol Biol. 2019. PMID: 31285605 Free PMC article.
-
Uniparental disomy unveils a novel recessive mutation in POMT2.Neuromuscul Disord. 2018 Jul;28(7):592-596. doi: 10.1016/j.nmd.2018.04.003. Epub 2018 Apr 10. Neuromuscul Disord. 2018. PMID: 29759639 Free PMC article.
References
-
- Kim DS, Hayashi YK, Matsumoto H, Ogawa M, Noguchi S, Murakami N, et al. POMT1 mutation results in defective glycosylation and loss of laminin-binding activity in alpha-DG. Neurology. 2004;62:1009–11. - PubMed
-
- Akasaka-Manya K, Manya H, Nakajima A, Kawakita M, Endo T. Physical and functional association of human protein O-mannosyltransferases 1 and 2. J Biol Chem. 2006;281:19339–45. - PubMed
-
- Mercuri E, D’Amico A, Tessa A, Berardinelli A, Pane M, Messina S, et al. POMT2 mutation in a patient with ’MEB-like’ phenotype. Neuromuscul Disord. 2006;16:446–8. - PubMed
-
- Yanagisawa A, Bouchet C, Van den Bergh PY, Cuisset JM, Viollet L, Leturcq F, et al. New POMT2 mutations causing congenital muscular dystrophy: identification of a founder mutation. Neurology. 2007;69:1254–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
