

Genotoxic stress-induced cyclin D1 phosphorylation and proteolysis are required for genomic stability
- PMID: 18809569
- PMCID: PMC2593367
- DOI: 10.1128/MCB.01085-08
Genotoxic stress-induced cyclin D1 phosphorylation and proteolysis are required for genomic stability
Abstract
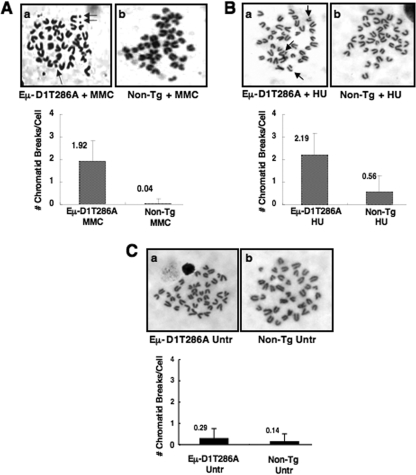
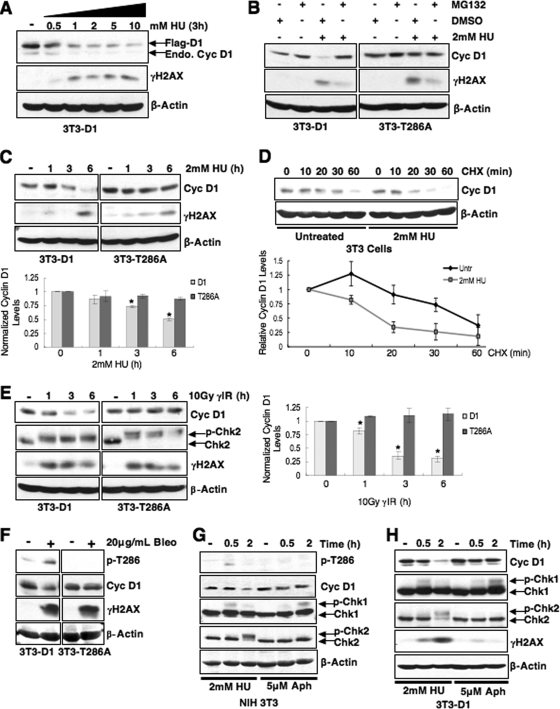
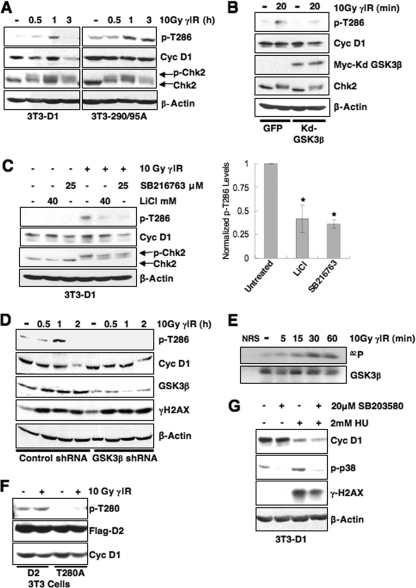
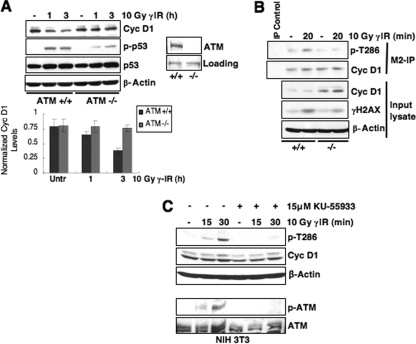
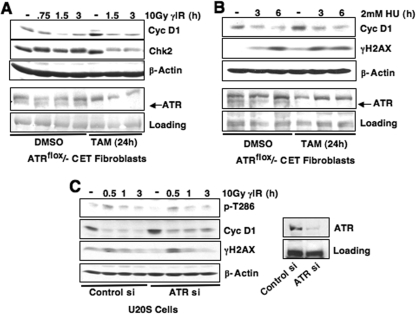
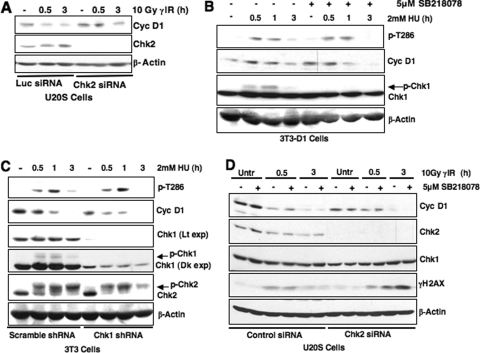
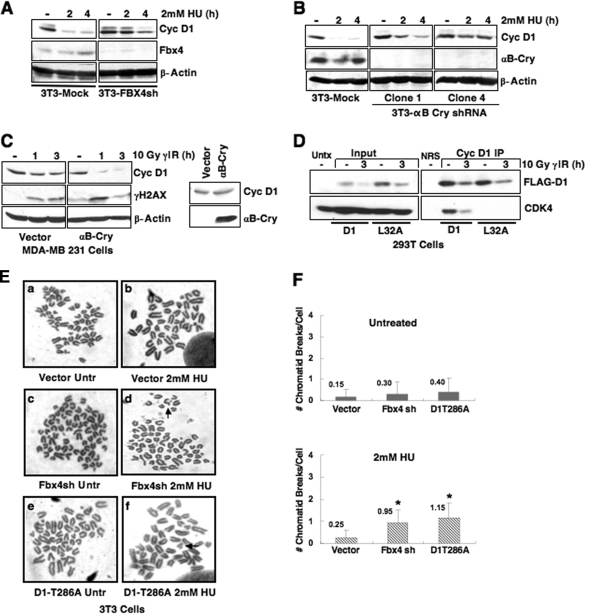
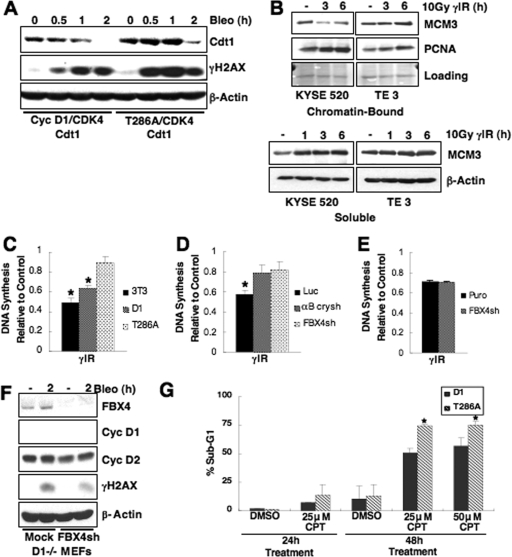
While mitogenic induction of cyclin D1 contributes to cell cycle progression, ubiquitin-mediated proteolysis buffers this accumulation and prevents aberrant proliferation. Because the failure to degrade cyclin D1 during S-phase triggers DNA rereplication, we have investigated cellular regulation of cyclin D1 following genotoxic stress. These data reveal that expression of cyclin D1 alleles refractory to phosphorylation- and ubiquitin-mediated degradation increase the frequency of chromatid breaks following DNA damage. Double-strand break-dependent cyclin D1 degradation requires ATM and GSK3beta, which in turn mediate cyclin D1 phosphorylation. Phosphorylated cyclin D1 is targeted for proteasomal degradation after ubiquitylation by SCF(Fbx4-alphaBcrystallin). Loss of Fbx4-dependent degradation triggers radio-resistant DNA synthesis, thereby sensitizing cells to S-phase-specific chemotherapeutic intervention. These data suggest that failure to degrade cyclin D1 compromises the intra-S-phase checkpoint and suggest that cyclin D1 degradation is a vital cellular response necessary to prevent genomic instability following genotoxic insult.
Figures

References
-
- Agami, R., and R. Bernards. 2000. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell 10255-66. - PubMed
-
- Aggarwal, P., M. D. Lessie, D. I. Lin, L. Pontano, A. B. Gladden, B. Nuskey, A. Goradia, M. A. Wasik, A. J. Klein-Szanto, A. K. Rustgi, C. H. Bassing, and J. A. Diehl. 2007. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev. 212908-2922. - PMC - PubMed
-
- Barnes, D. M., and C. E. Gillett. 1998. Cyclin D1 in breast cancer. Breast Cancer Res. Treat. 521-15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous