

Dissection of the molecular defects caused by pathogenic mutations in the DNA repair factor XPC
- PMID: 18809580
- PMCID: PMC2593387
- DOI: 10.1128/MCB.00781-08
Dissection of the molecular defects caused by pathogenic mutations in the DNA repair factor XPC
Abstract
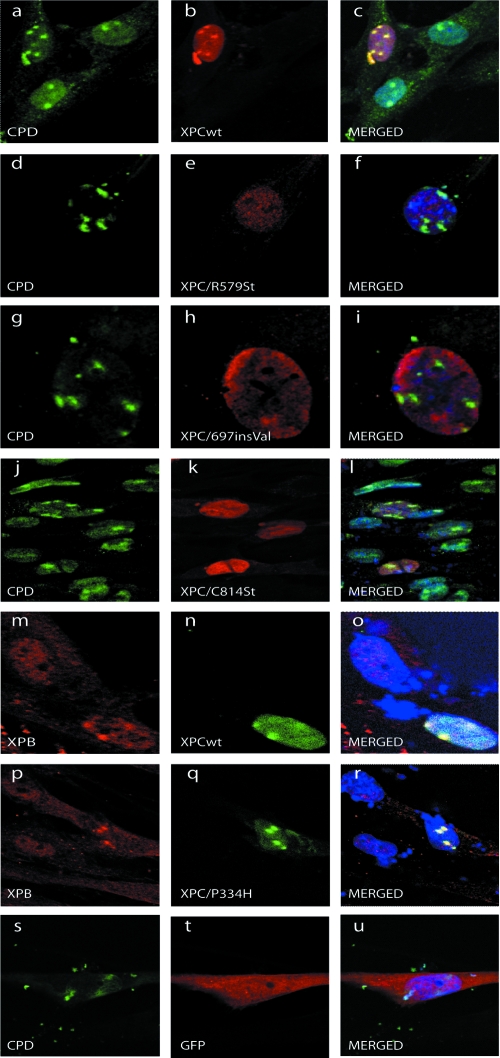
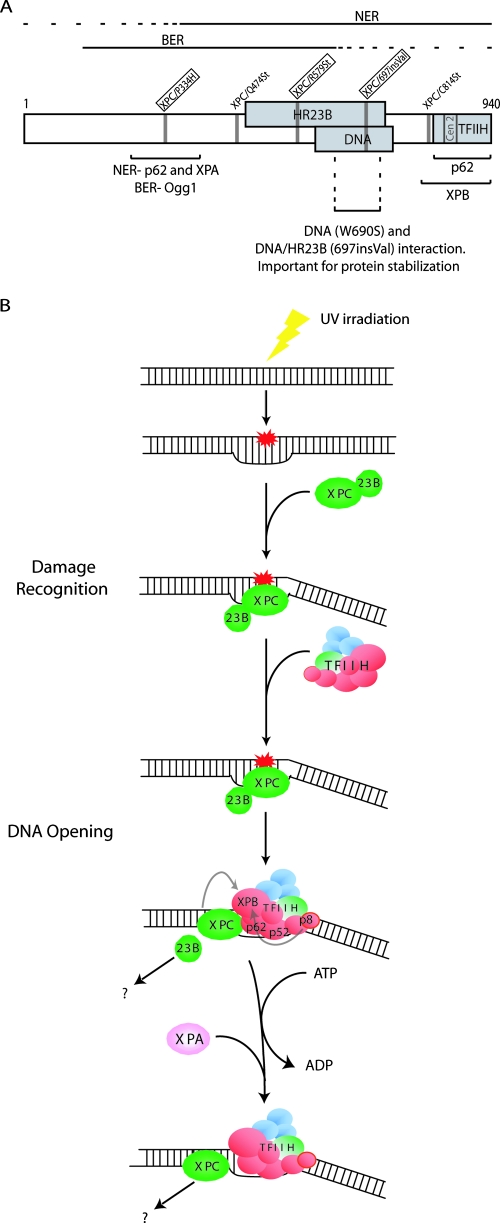
XPC is responsible for DNA damage sensing in nucleotide excision repair (NER). Mutations in XPC lead to a defect in NER and to xeroderma pigmentosum (XP-C). Here, we analyzed the biochemical properties behind mutations found within three patients: one amino acid substitution (P334H, XP1MI, and GM02096), one amino acid incorporation in a conserved domain (697insVal, XP8BE, and GM02249), and a stop mutation (R579St, XP67TMA, and GM14867). Using these mutants, we demonstrated that HR23B stabilizes XPC on DNA and protects it from degradation. XPC recruits the transcription/repair factor TFIIH and stimulates its XPB ATPase activity to initiate damaged DNA opening. In an effort to understand the severity of XP-C phenotypes, we also demonstrated that single mutations in XPC perturb other repair processes, such as base excision repair (e.g., the P334H mutation prevents the stimulation of Ogg1 glycosylase because it thwarts the interaction between XPC and Ogg1), thereby leading to a deeper understanding of the molecular repair defect of the XP-C patients.
Figures
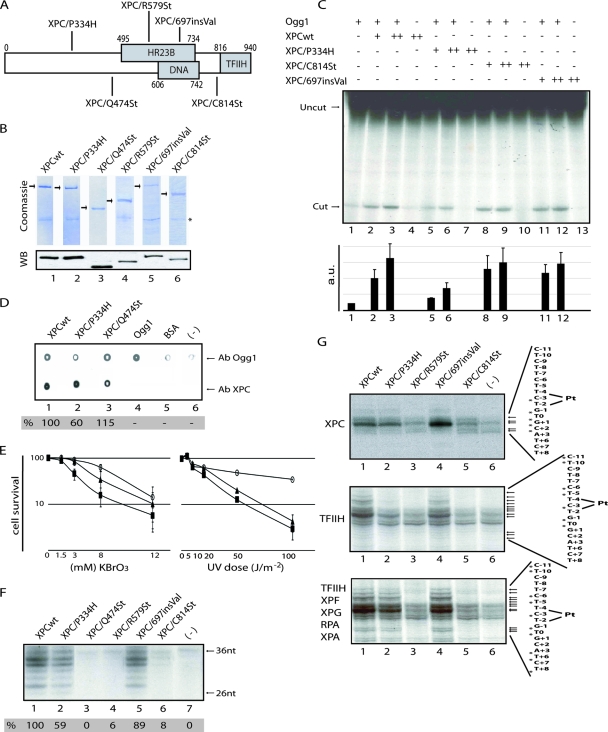
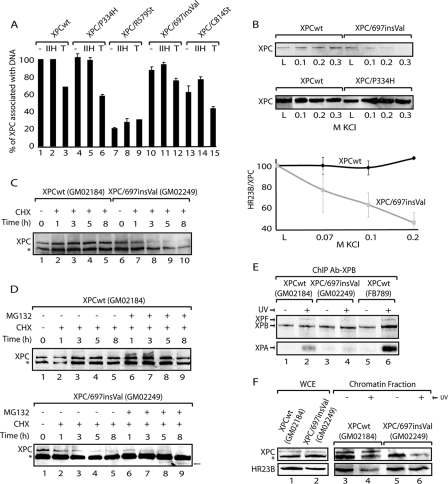
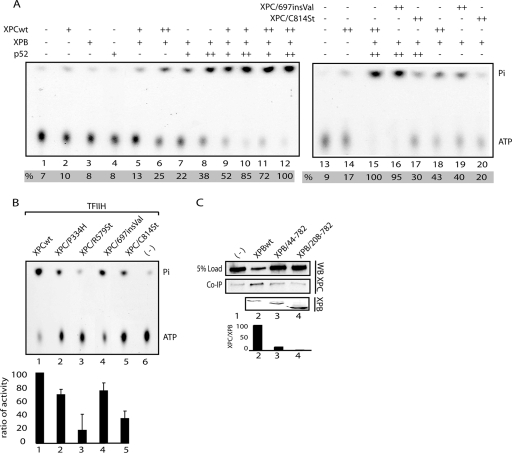
 ), when washed with 0.07 to 0.2 M KCl as indicated (lower graph). Error bars correspondent to the standard error of the mean of two independent experiments. (C) GM02184 (XPCwt) or GM02249 (XPC/697insVal) human lymphoblasts were incubated overtime with 0.1 mM CHX. Cell extracts were then analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting with an XPC antibody. The asterisk corresponds to a nonspecific band. (D) GM02184 (XPCwt) or GM02249 (XPC/697insVal) human lymphoblasts were incubated during the indicated times with CHX either alone or in combination with MG132. Cell extracts were analyzed as described in panel C. The arrow indicates a probable degradation product (55). (E) ChIP followed by Western blot analysis of Ab-XPB immunoprecipitated samples from GM02184 (XPCwt), GM02249 (XPC/697insVal), and FB789 (XPCwt) cell lines fixed at t = 0 (no UV) or t = 15 min after UV irradiation (20 J/m2). A total of 400 μg of formaldehyde cross-linked protein extract was used per immunoprecipitation. (F) Portions (100 μg) of whole-cell extracts (WCE) or 40 μg of chromatin fraction extract from GM02184 or GM02249 cells were immunoblotted for the presence of XPC and HR23B. The asterisk corresponds to a nonspecific band.
), when washed with 0.07 to 0.2 M KCl as indicated (lower graph). Error bars correspondent to the standard error of the mean of two independent experiments. (C) GM02184 (XPCwt) or GM02249 (XPC/697insVal) human lymphoblasts were incubated overtime with 0.1 mM CHX. Cell extracts were then analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting with an XPC antibody. The asterisk corresponds to a nonspecific band. (D) GM02184 (XPCwt) or GM02249 (XPC/697insVal) human lymphoblasts were incubated during the indicated times with CHX either alone or in combination with MG132. Cell extracts were analyzed as described in panel C. The arrow indicates a probable degradation product (55). (E) ChIP followed by Western blot analysis of Ab-XPB immunoprecipitated samples from GM02184 (XPCwt), GM02249 (XPC/697insVal), and FB789 (XPCwt) cell lines fixed at t = 0 (no UV) or t = 15 min after UV irradiation (20 J/m2). A total of 400 μg of formaldehyde cross-linked protein extract was used per immunoprecipitation. (F) Portions (100 μg) of whole-cell extracts (WCE) or 40 μg of chromatin fraction extract from GM02184 or GM02249 cells were immunoblotted for the presence of XPC and HR23B. The asterisk corresponds to a nonspecific band.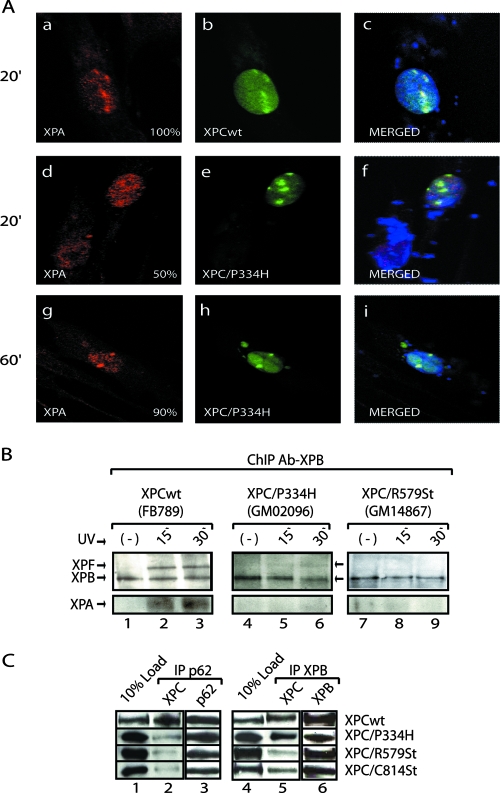
References
-
- Aburatani, H., Y. Hippo, T. Ishida, R. Takashima, C. Matsuba, T. Kodama, M. Takao, A. Yasui, K. Yamamoto, and M. Asano. 1997. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res. 572151-2156. - PubMed
-
- Bootsma, D., K. H. Kraemer, J. E. Cleaver, and J. H. J. Hoeijmakers. 2002. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy, p. 211-237. In B. Vogelstein and K. W. Kinzler (ed.), The genetic basis of human cancer, 2nd ed. McGraw-Hill, New York, NY.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials