

Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction
- PMID: 18809807
- PMCID: PMC2756193
- DOI: 10.1161/CIRCULATIONAHA.108.784298
Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction
Abstract
Background: Oxidative injury and dysfunction of the vascular endothelium are early and causal features of many vascular diseases. Single antioxidant strategies to prevent vascular injury have met with mixed results.
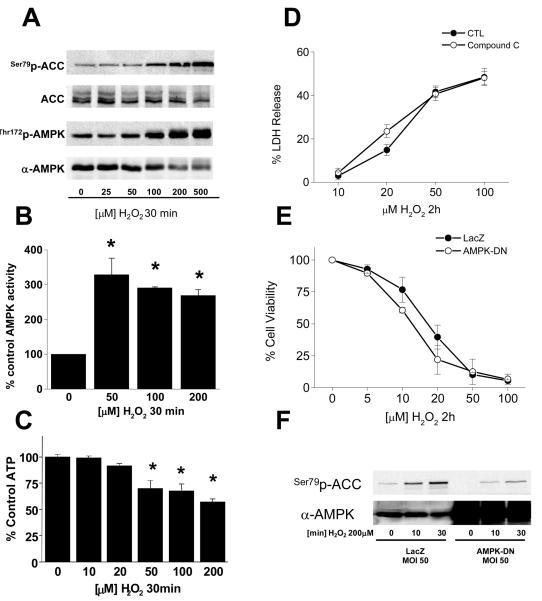
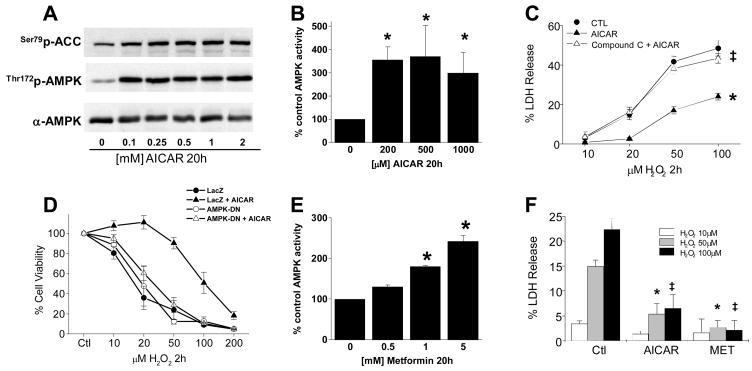
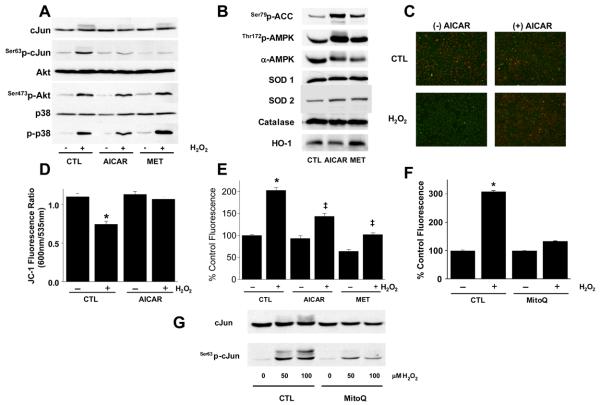
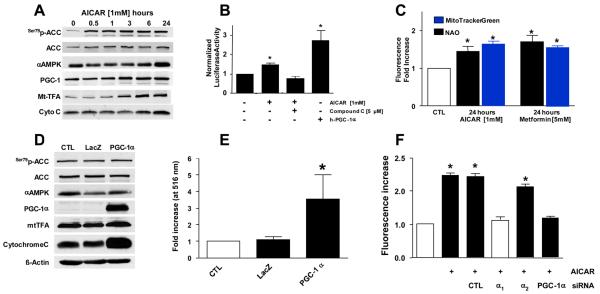
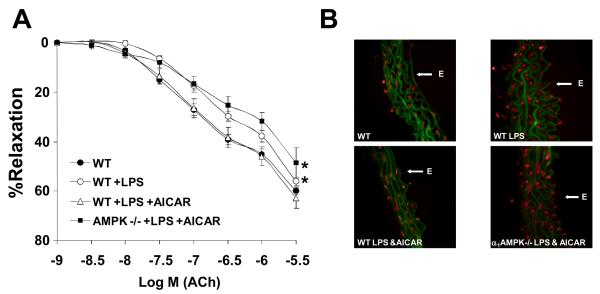
Methods and results: Here, we report that induction of a metabolic stress response with adenosine monophosphate kinase (AMPK) prevents oxidative endothelial cell injury. This response is characterized by stabilization of the mitochondrion and increased mitochondrial biogenesis, resulting in attenuation of oxidative c-Jun N-terminal kinase (JNK) activation. We report that peroxisome proliferator coactivator 1alpha is a key downstream target of AMPK that is both necessary and sufficient for the metabolic stress response and JNK attenuation. Moreover, induction of the metabolic stress response in vivo attenuates reactive oxygen species-mediated JNK activation and endothelial dysfunction in response to angiotensin II in wild-type mice but not in animals lacking either the endothelial isoform of AMPK or peroxisome proliferator coactivator 1alpha.
Conclusions: These data highlight AMPK and peroxisome proliferator coactivator 1alpha as potential therapeutic targets for the amelioration of endothelial dysfunction and, as a consequence, vascular disease.
Figures


Similar articles
-
Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells.Diabetes. 2006 Jan;55(1):120-7. Diabetes. 2006. PMID: 16380484
-
PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells.Cardiovasc Res. 2005 Jun 1;66(3):562-73. doi: 10.1016/j.cardiores.2005.01.026. Epub 2005 Feb 25. Cardiovasc Res. 2005. PMID: 15914121
-
Age-dependent cardiac function during experimental sepsis: effect of pharmacological activation of AMP-activated protein kinase by AICAR.Am J Physiol Heart Circ Physiol. 2018 Oct 1;315(4):H826-H837. doi: 10.1152/ajpheart.00052.2018. Epub 2018 Jul 6. Am J Physiol Heart Circ Physiol. 2018. PMID: 29979626 Free PMC article.
-
Adenosine monophosphate activated protein kinase regulates ABCG1-mediated oxysterol efflux from endothelial cells and protects against hypercholesterolemia-induced endothelial dysfunction.Arterioscler Thromb Vasc Biol. 2010 Jul;30(7):1354-62. doi: 10.1161/ATVBAHA.110.204230. Epub 2010 Apr 15. Arterioscler Thromb Vasc Biol. 2010. PMID: 20395595
-
The AMPK-PGC-1α signaling axis regulates the astrocyte glutathione system to protect against oxidative and metabolic injury.Neurobiol Dis. 2018 May;113:59-69. doi: 10.1016/j.nbd.2018.02.004. Epub 2018 Feb 10. Neurobiol Dis. 2018. PMID: 29438738
Cited by
-
High fat diet enhances cardiac abnormalities in SHR rats: Protective role of heme oxygenase-adiponectin axis.Diabetol Metab Syndr. 2011 Dec 23;3(1):37. doi: 10.1186/1758-5996-3-37. Diabetol Metab Syndr. 2011. PMID: 22196253 Free PMC article.
-
Influence of mental stress and environmental toxins on circadian clocks: Implications for redox regulation of the heart and cardioprotection.Br J Pharmacol. 2020 Dec;177(23):5393-5412. doi: 10.1111/bph.14949. Epub 2020 Feb 4. Br J Pharmacol. 2020. PMID: 31833063 Free PMC article. Review.
-
Adipocyte heme oxygenase-1 induction attenuates metabolic syndrome in both male and female obese mice.Hypertension. 2010 Dec;56(6):1124-30. doi: 10.1161/HYPERTENSIONAHA.110.151423. Epub 2010 Nov 1. Hypertension. 2010. PMID: 21041703 Free PMC article.
-
Mechanisms of vascular aging: new perspectives.J Gerontol A Biol Sci Med Sci. 2010 Oct;65(10):1028-41. doi: 10.1093/gerona/glq113. Epub 2010 Jun 24. J Gerontol A Biol Sci Med Sci. 2010. PMID: 20576649 Free PMC article. Review.
-
Cognitive Decline, Dementia, Alzheimer's Disease and Presbycusis: Examination of the Possible Molecular Mechanism.Front Neurosci. 2018 Jun 8;12:394. doi: 10.3389/fnins.2018.00394. eCollection 2018. Front Neurosci. 2018. PMID: 29937713 Free PMC article. Review.
References
-
- Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007 October;7(10):803–15. - PubMed
-
- Widlansky ME, Gokce N, Keaney JF, Jr., Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003 October 1;42(7):1149–60. - PubMed
-
- Gokce N, Keaney JF, Jr., Hunter LM, et al. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events inpatients with peripheral vascular disease. J Am Coll Cardiol. 2003 May 21;41(10):1769–75. - PubMed
-
- Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000 January 20;342(3):154–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
