

doi: 10.1007/s11568-008-9026-9.
Epub 2008 Sep 20.
Two sisters with Rett syndrome and non-identical paternally-derived microdeletions in the MECP2 gene
Affiliations
- PMID: 18810657
- PMCID: PMC2694857
- DOI: 10.1007/s11568-008-9026-9
Item in Clipboard
Two sisters with Rett syndrome and non-identical paternally-derived microdeletions in the MECP2 gene
Genomic Med.
2008 Dec.
Abstract
The unique case of two sisters with symptoms of RTT and two quite distinct, novel, and apparently de novo microdeletions of the MECP2 gene is described. One sister possessed an 18 base-pair (bp) deletion (c.1155_1172del18) within the deletion hotspot region of exon 4, whereas the other sister exhibited a 43 bp deletion at a different location in the same exon (c.1448_1461del14+29). Although these lesions occurred on the same paternally-derived X chromosome, this is probably due to chance co-occurrence owing to the relatively high mutation rate of the MECP2 gene rather than to a constitutional mutator phenotype.
Figures

Pedigree of the affected family; The clinically affected sisters (Cases 1 and 2) are denoted by filled circles. The MECP2 genotypes of the sisters are given. The paternal inheritance of the c.1373G>A transition is indicated
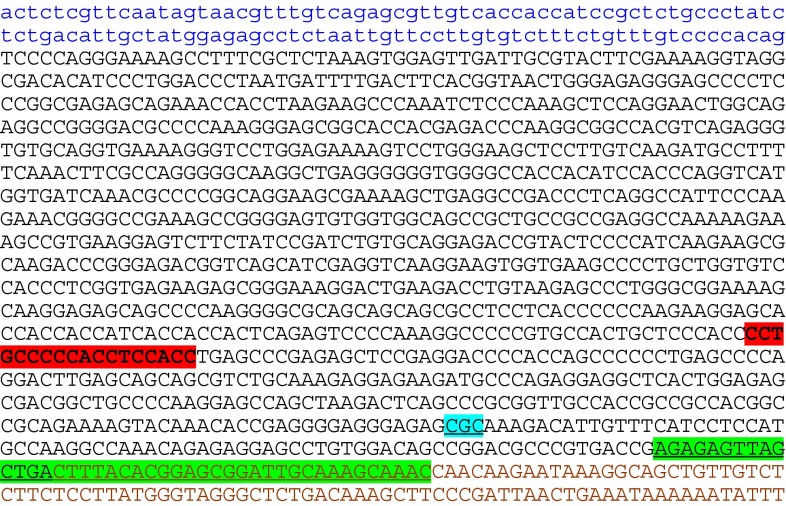
Sequence of exon 4 of the MECP2 gene showing the locations of the two microdeletions and the missense mutation detected in the reported family
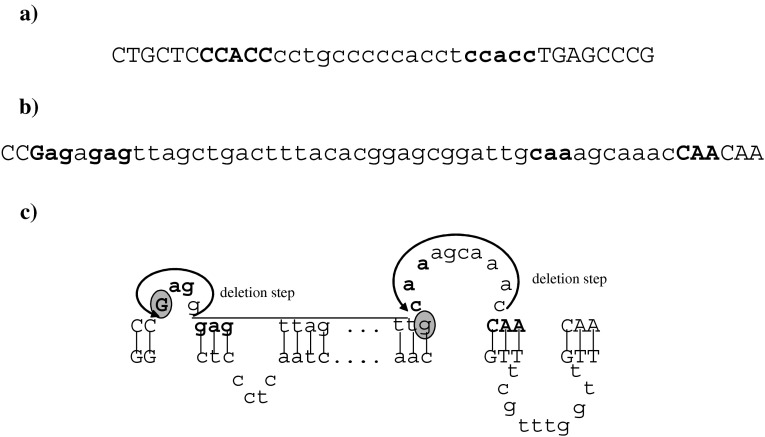
Repetitive sequence elements in the vicinity of the microdeletion breakpoints and the secondary structures postulated to have been involved in the genesis of the two mutations. Lower case letters denote deleted nucleotides. (a) Repetitive sequence elements (shown in bold) found in the vicinity of the breakpoints of the 1155_1172del18 microdeletion. (b) Repetitive sequence elements (shown in bold) found in the vicinity of the breakpoints of the 1448_1461del14+29 microdeletion. (c) Schematic representation of the postulated non-B DNA slipped structures at the breakpoint of the 1448_1461del14+29 microdeletion. The nucleotides shown in bold type correspond to the direct repeats shown in (b). Nucleotides circled in grey denote the homology between two breakpoint junctions. The arrows indicate the sequences deleted
Similar articles
-
[Analysis of the parental origin of de novo MECP2 mutations and X chromosome inactivation in fifteen sporadic cases with Rett syndrome].Zhonghua Er Ke Za Zhi. 2009 Aug;47(8):565-9. Zhonghua Er Ke Za Zhi. 2009. PMID: 19951486 Chinese.
-
MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin.Am J Hum Genet. 2001 May;68(5):1093-101. doi: 10.1086/320109. Epub 2001 Apr 17. Am J Hum Genet. 2001. PMID: 11309679 Free PMC article.
-
Two novel mutations in the MECP2 gene in patients with Rett syndrome.Gene. 2020 Mar 30;732:144337. doi: 10.1016/j.gene.2020.144337. Epub 2020 Jan 17. Gene. 2020. PMID: 31958484
-
Exploring the possible link between MeCP2 and oxidative stress in Rett syndrome.Free Radic Biol Med. 2015 Nov;88(Pt A):81-90. doi: 10.1016/j.freeradbiomed.2015.04.019. Epub 2015 May 8. Free Radic Biol Med. 2015. PMID: 25960047 Review.
-
Rett syndrome: from the gene to the disease.Eur Neurol. 2009;61(1):3-10. doi: 10.1159/000165342. Epub 2008 Oct 24. Eur Neurol. 2009. PMID: 18948693 Review.
Cited by
-
Monozygotic twins with Rett syndrome: Phenotyping the first two years of life.J Dev Phys Disabil. 2014 Apr;26(2):171-182. doi: 10.1007/s10882-013-9351-3. J Dev Phys Disabil. 2014. PMID: 29769795 Free PMC article.
References
-
- Archer H, Evans J, Leonard H, Colvin L, Ravine D, Christodoulou J, Williamson S, Charman T, Bailey MES, Sampson J, de Klerk N, Clarke A. Correlation between clinical severity in patients with Rett syndrome with a p.R168X or p.T158M MECP2 mutation, and the direction and degree of skewing of X-chromosome inactivation. J Med Genet. 2007;44:148–152. doi: 10.1136/jmg.2006.045260. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources