

Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration
- PMID: 18816186
- PMCID: PMC2713807
- DOI: 10.1089/ars.2008.2130
Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration
Abstract
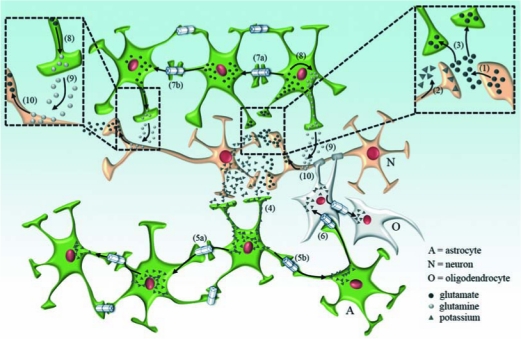
In normal brain, neurons, astrocytes, and oligodendrocytes, the most abundant and active cells express pannexins and connexins, protein subunits of two families forming membrane channels. Most available evidence indicates that in mammals endogenously expressed pannexins form only hemichannels and connexins form both gap junction channels and hemichannels. Whereas gap junction channels connect the cytoplasm of contacting cells and coordinate electric and metabolic activity, hemichannels communicate the intra- and extracellular compartments and serve as a diffusional pathway for ions and small molecules. A subthreshold stimulation by acute pathological threatening conditions (e.g., global ischemia subthreshold for cell death) enhances neuronal Cx36 and glial Cx43 hemichannel activity, favoring ATP release and generation of preconditioning. If the stimulus is sufficiently deleterious, microglia become overactivated and release bioactive molecules that increase the activity of hemichannels and reduce gap junctional communication in astroglial networks, depriving neurons of astrocytic protective functions, and further reducing neuronal viability. Continuous glial activation triggered by low levels of anomalous proteins expressed in several neurodegenerative diseases induce glial hemichannel and gap junction channel disorders similar to those of acute inflammatory responses triggered by ischemia or infectious diseases. These changes are likely to occur in diverse cell types of the CNS and contribute to neurodegeneration during inflammatory process.
Figures







References
-
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. - PubMed
-
- Aguado F. Ballabriga J. Pozas E. Ferrer I. TrkA immunoreactivity in reactive astrocytes in human neurodegenerative diseases and colchicine-treated rats. Acta Neuropathol. 1998;96:495–501. - PubMed
-
- Ahmad AS. Zhuang H. Echeverria V. Dore S. Stimulation of prostaglandin EP2 receptors prevents NMDA-induced excitotoxicity. J Neurotrauma. 2006;23:1895–1903. - PubMed
-
- Aizenman E. Lipton SA. Loring RH. Selective modulation of NMDA responses by reduction and oxidation. Neuron. 1989;2:1257–1263. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
