

Precision proteomics: the case for high resolution and high mass accuracy
- PMID: 18818311
- PMCID: PMC2587563
- DOI: 10.1073/pnas.0800788105
Precision proteomics: the case for high resolution and high mass accuracy
Abstract
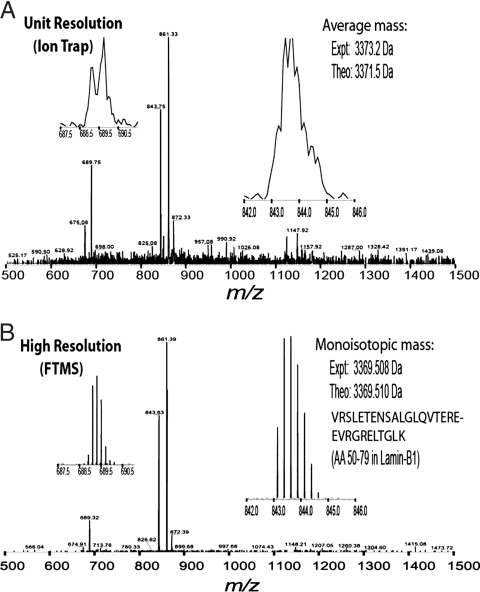
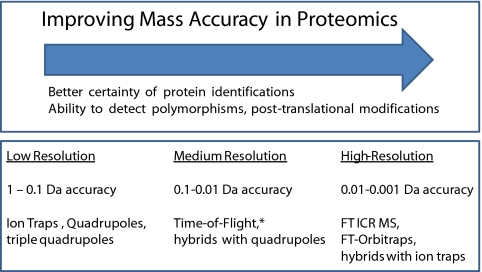
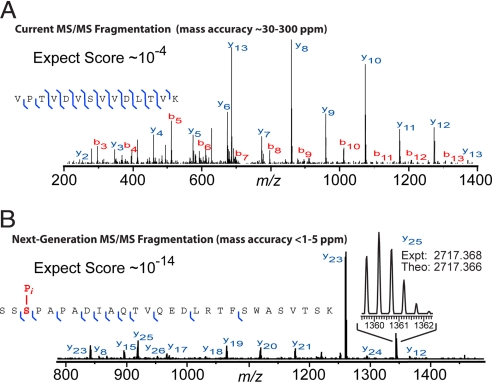
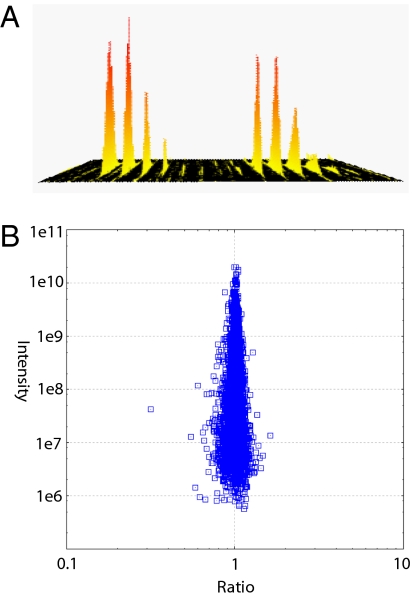
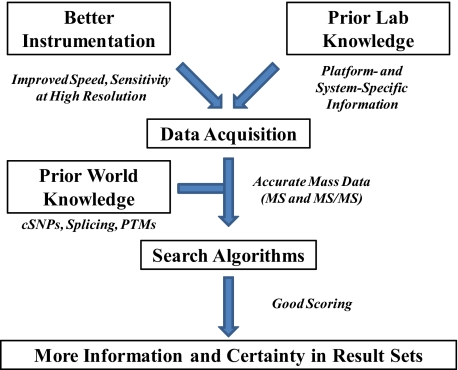
Proteomics has progressed radically in the last 5 years and is now on par with most genomic technologies in throughput and comprehensiveness. Analyzing peptide mixtures by liquid chromatography coupled to high-resolution mass spectrometry (LC-MS) has emerged as the main technology for in-depth proteome analysis whereas two-dimensional gel electrophoresis, low-resolution MALDI, and protein arrays are playing niche roles. MS-based proteomics is rapidly becoming quantitative through both label-free and stable isotope labeling technologies. The latest generation of mass spectrometers combines extremely high resolving power, mass accuracy, and very high sequencing speed in routine proteomic applications. Peptide fragmentation is mostly performed in low-resolution but very sensitive and fast linear ion traps. However, alternative fragmentation methods and high-resolution fragment analysis are becoming much more practical. Recent advances in computational proteomics are removing the data analysis bottleneck. Thus, in a few specialized laboratories, "precision proteomics" can now identify and quantify almost all fragmented peptide peaks. Huge challenges and opportunities remain in technology development for proteomics; thus, this is not "the beginning of the end" but surely "the end of the beginning."
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Fountoulakis M, Tsangaris G, Oh JE, Maris A, Lubec G. Protein profile of the HeLa cell line. J Chromatogr A. 2004;1038:247–265. - PubMed
-
- Campostrini N, et al. Spot overlapping in two-dimensional maps: A serious problem ignored for much too long. Proteomics. 2005;5:2385–2395. - PubMed
-
- Petrak J, et al. Deja vu in proteomics. A hit parade of repeatedly identified differentially expressed proteins. Proteomics. 2008;8:1744–1749. - PubMed
-
- Angenendt P, Kreutzberger J, Glokler J, Hoheisel JD. Generation of high density protein microarrays by cell-free in situ expression of unpurified PCR products. Mol Cell Proteomics. 2006;5:1658–1666. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
