

Cell-restricted immortalization by human papillomavirus correlates with telomerase activation and engagement of the hTERT promoter by Myc
- PMID: 18818322
- PMCID: PMC2583678
- DOI: 10.1128/JVI.01318-08
Cell-restricted immortalization by human papillomavirus correlates with telomerase activation and engagement of the hTERT promoter by Myc
Abstract
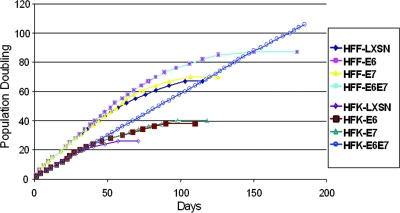
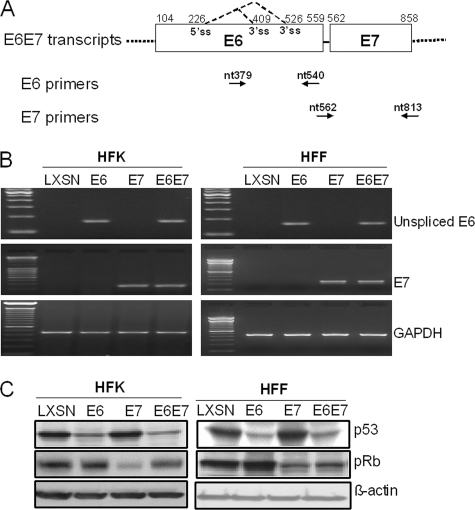
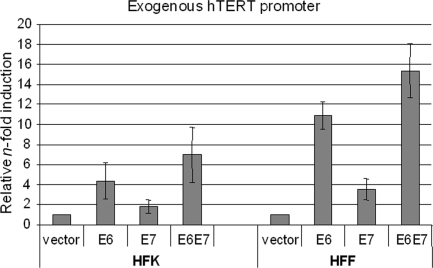
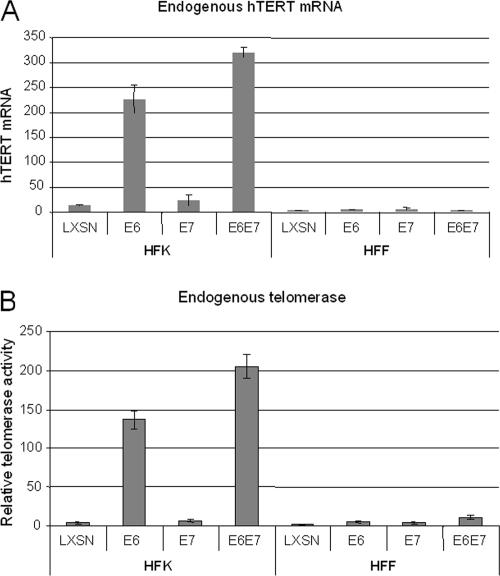
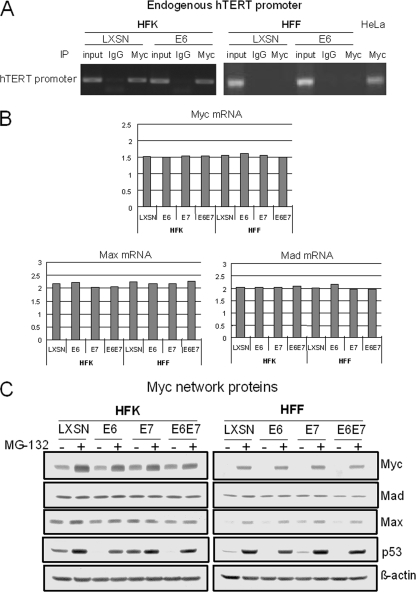
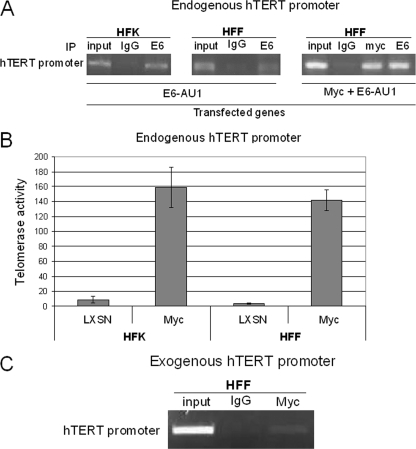
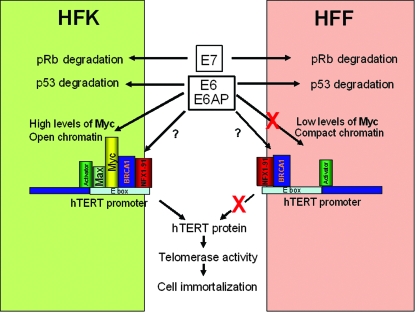
The high-risk human papillomaviruses (HPVs) are the causative agents of nearly all cervical cancers and are etiologically linked to additional human cancers, including those of anal, oral, and laryngeal origin. The main transforming genes of the high-risk HPVs are E6 and E7. E6, in addition to its role in p53 degradation, induces hTERT mRNA transcription in genital keratinocytes via interactions with Myc protein, thereby increasing cellular telomerase activity. While the HPV type 16 E6 and E7 genes efficiently immortalize human keratinocytes, they appear to only prolong the life span of human fibroblasts. To examine the molecular basis for this cell-type dependency, we examined the correlation between the ability of E6 to transactivate endogenous and exogenous hTERT promoters and to immortalize genital keratinocytes and fibroblasts. Confirming earlier studies, the E6 and E7 genes were incapable of immortalizing human fibroblasts but did delay senescence. Despite the lack of immortalization, E6 was functional in the fibroblasts, mediating p53 degradation and strongly transactivating an exogenous hTERT promoter. However, E6 failed to transactivate the endogenous hTERT promoter. Coordinately with this failure, we observed that Myc protein was not associated with the endogenous hTERT promoter, most likely due to the extremely low level of Myc expression in these cells and/or to differences in chromatin structure, in contrast with hTERT promoters that we found to be activated by E6 (i.e., the endogenous hTERT promoter in primary keratinoctyes and the exogenous hTERT core promoter in fibroblasts), where Myc is associated with the promoter in either a quiescent or an E6-induced state. These findings are consistent with those of our previous studies on mutagenesis and the knockdown of small interfering RNA, which demonstrated a requirement for Myc in the induction of the hTERT promoter by E6 and suggested that occupancy of the promoter by Myc determines the responsiveness of E6 and the downstream induction of telomerase and cell immortalization.
Figures
References
-
- Banks, L., P. Spence, E. Androphy, N. Hubbert, G. Matlashewski, A. Murray, and L. Crawford. 1987. Identification of human papillomavirus type 18 E6 polypeptide in cells derived from human cervical carcinomas. J. Gen. Virol. 681351-1359. - PubMed
-
- Benanti, J. A., M. L. Wang, H. E. Myers, K. L. Robinson, C. Grandori, and D. A. Galloway. 2007. Epigenetic down-regulation of ARF expression is a selection step in immortalization of human fibroblasts by c-Myc. Mol. Cancer Res. 51181-1189. - PubMed
-
- Casillas, M. A., S. L. Brotherton, L. G. Andrews, J. M. Ruppert, and T. O. Tollefsbol. 2003. Induction of endogenous telomerase (hTERT) by c-Myc in WI-38 fibroblasts transformed with specific genetic elements. Gene 31657-65. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
