

Sequencing of natural strains of Arabidopsis thaliana with short reads
- PMID: 18818371
- PMCID: PMC2593571
- DOI: 10.1101/gr.080200.108
Sequencing of natural strains of Arabidopsis thaliana with short reads
Abstract
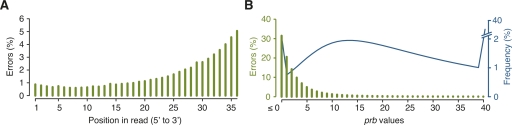
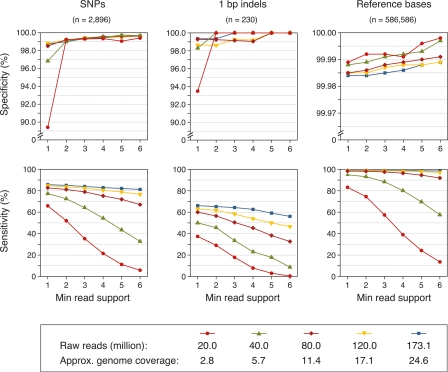
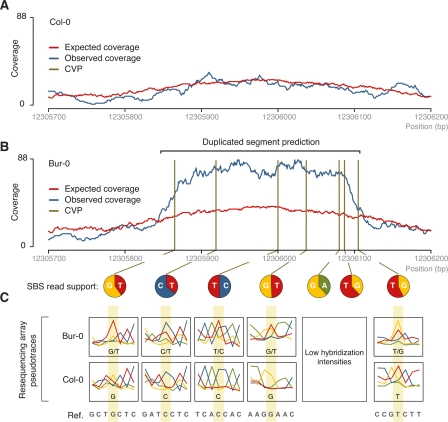
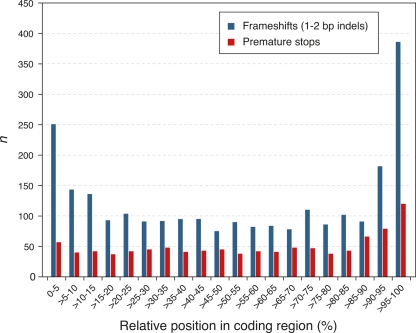
Whole-genome hybridization studies have suggested that the nuclear genomes of accessions (natural strains) of Arabidopsis thaliana can differ by several percent of their sequence. To examine this variation, and as a first step in the 1001 Genomes Project for this species, we produced 15- to 25-fold coverage in Illumina sequencing-by-synthesis (SBS) reads for the reference accession, Col-0, and two divergent strains, Bur-0 and Tsu-1. We aligned reads to the reference genome sequence to assess data quality metrics and to detect polymorphisms. Alignments revealed 823,325 unique single nucleotide polymorphisms (SNPs) and 79,961 unique 1- to 3-bp indels in the divergent accessions at a specificity of >99%, and over 2000 potential errors in the reference genome sequence. We also identified >3.4 Mb of the Bur-0 and Tsu-1 genomes as being either extremely dissimilar, deleted, or duplicated relative to the reference genome. To obtain sequences for these regions, we incorporated the Velvet assembler into a targeted de novo assembly method. This approach yielded 10,921 high-confidence contigs that were anchored to flanking sequences and harbored indels as large as 641 bp. Our methods are broadly applicable for polymorphism discovery in moderate to large genomes even at highly diverged loci, and we established by subsampling the Illumina SBS coverage depth required to inform a broad range of functional and evolutionary studies. Our pipeline for aligning reads and predicting SNPs and indels, SHORE, is available for download at http://1001genomes.org.
Figures

References
-
- The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous