

SLEPR: a sample-level enrichment-based pathway ranking method -- seeking biological themes through pathway-level consistency
- PMID: 18818771
- PMCID: PMC2546449
- DOI: 10.1371/journal.pone.0003288
SLEPR: a sample-level enrichment-based pathway ranking method -- seeking biological themes through pathway-level consistency
Abstract
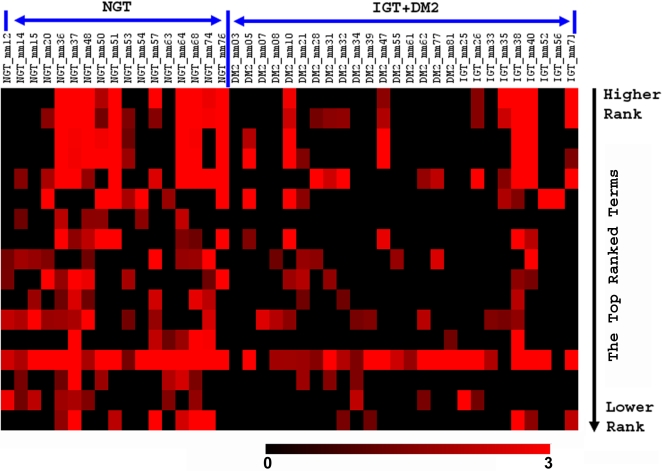
Analysis of microarray and other high throughput data often involves identification of genes consistently up or down-regulated across samples as the first step in extraction of biological meaning. This gene-level paradigm can be limited as a result of valid sample fluctuations and biological complexities. In this report, we describe a novel method, SLEPR, which eliminates this limitation by relying on pathway-level consistencies. Our method first selects the sample-level differentiated genes from each individual sample, capturing genes missed by other analysis methods, ascertains the enrichment levels of associated pathways from each of those lists, and then ranks annotated pathways based on the consistency of enrichment levels of individual samples from both sample classes. As a proof of concept, we have used this method to analyze three public microarray datasets with a direct comparison with the GSEA method, one of the most popular pathway-level analysis methods in the field. We found that our method was able to reproduce the earlier observations with significant improvements in depth of coverage for validated or expected biological themes, but also produced additional insights that make biological sense. This new method extends existing analyses approaches and facilitates integration of different types of HTP data.
Conflict of interest statement
Figures


References
-
- Hartigan JA, Wong MA. A k-means clustering algorithm. Applied Statistics. 1979;28:100–108.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
