

Role of microglial IKKbeta in kainic acid-induced hippocampal neuronal cell death
- PMID: 18819987
- PMCID: PMC2577806
- DOI: 10.1093/brain/awn230
Role of microglial IKKbeta in kainic acid-induced hippocampal neuronal cell death
Abstract
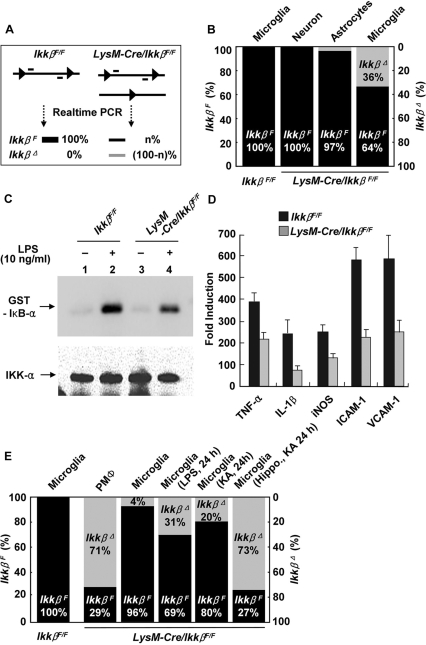
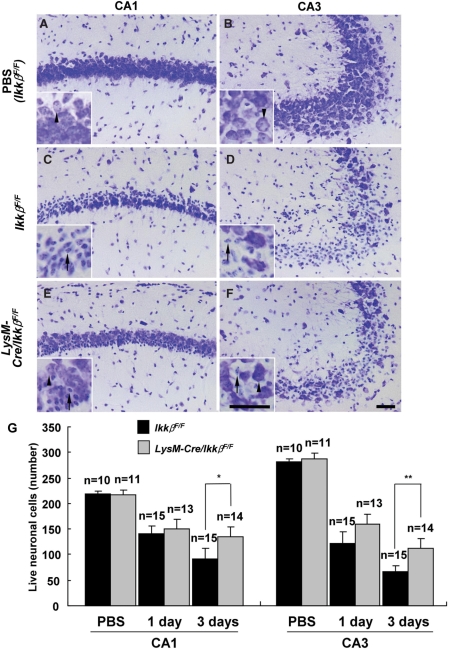
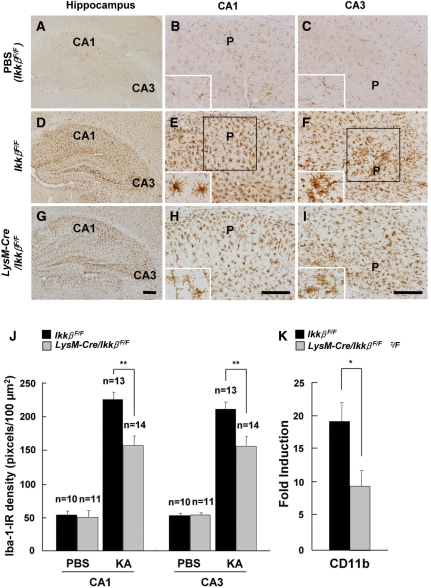
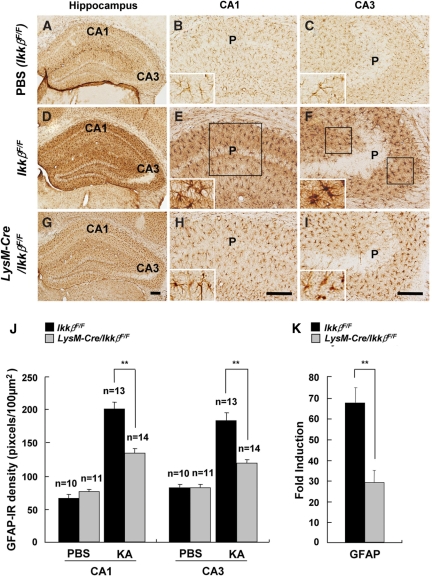
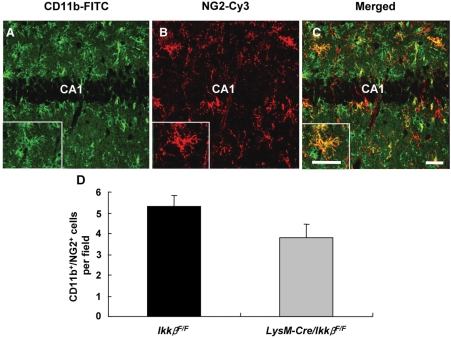
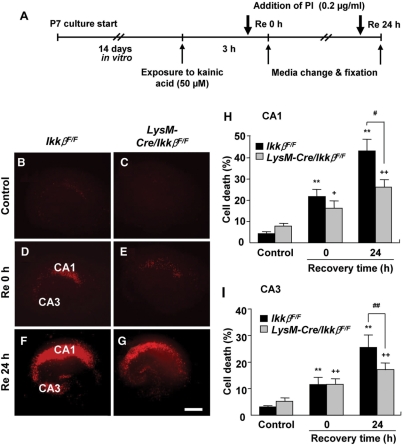
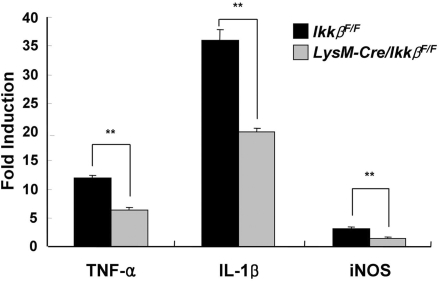
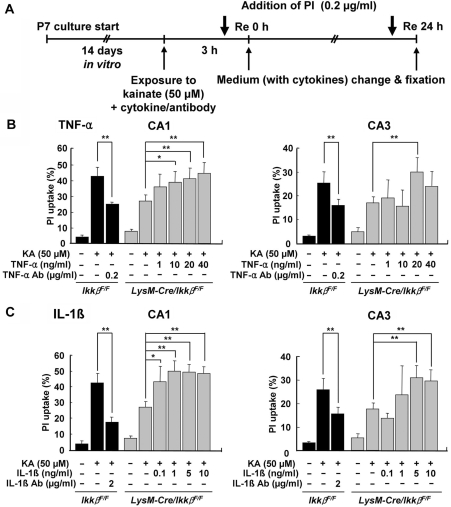
Microglial cells are activated during excitotoxin-induced neurodegeneration. However, the in vivo role of microglia activation in neurodegeneration has not yet been fully elucidated. To this end, we used Ikkbeta conditional knockout mice (LysM-Cre/Ikkbeta(F/F)) in which the Ikkbeta gene is specifically deleted in cells of myeloid lineage, including microglia, in the CNS. This deletion reduced IkappaB kinase (IKK) activity in cultured primary microglia by up to 40% compared with wild-type (Ikkbeta(F/F)), and lipopolysaccharide-induced proinflammatory gene expression was also compromised. Kainic acid (KA)-induced hippocampal neuronal cell death was reduced by 30% in LysM-Cre/Ikkbeta(F/F) mice compared with wild-type mice. Reduced neuronal cell death was accompanied by decreased KA-induced glial cell activation and subsequent expression of proinflammatory genes such as tumour necrosis factor (TNF)-alpha and interleukin (IL)-1beta. Similarly, neurons in organotypic hippocampal slice cultures (OHSCs) from LysM-Cre/Ikkbeta(F/F) mouse brain were less susceptible to KA-induced excitotoxicity compared with wild-type OHSCs, due in part to decreased TNF-alpha and IL-1beta expression. Based on these data, we concluded that IKK/nuclear factor-kappaB dependent microglia activation contributes to KA-induced hippocampal neuronal cell death in vivo through induction of inflammatory mediators.
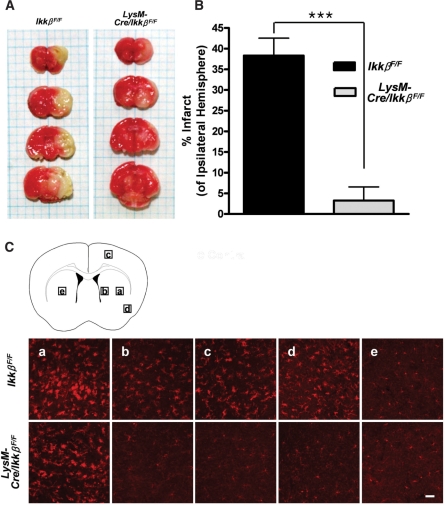
Figures
References
-
- Acarin L, Gonzalez B, Castellano B. STAT3 and NFkappaB activation precedes glial reactivity in the excitotoxically injured young cortex but not in the corresponding distal thalamic nuclei. J Neuropathol Exp Neurol. 2000;59:151–63. - PubMed
-
- Andersson PB, Perry VH, Gordon S. The CNS acute inflammatory response to excitotoxic neuronal cell death. Immunol Lett. 1991a;30:177–81. - PubMed
-
- Andersson PB, Perry VH, Gordon S. The kinetics and morphological characteristics of the macrophage-microglial response to kainic acid-induced neuronal degeneration. Neuroscience. 1991b;42:201–14. - PubMed
-
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–34. - PubMed
-
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
