

Molecular biology of bcr-abl1-positive chronic myeloid leukemia
- PMID: 18827185
- PMCID: PMC3952549
- DOI: 10.1182/blood-2008-03-144790
Molecular biology of bcr-abl1-positive chronic myeloid leukemia
Abstract
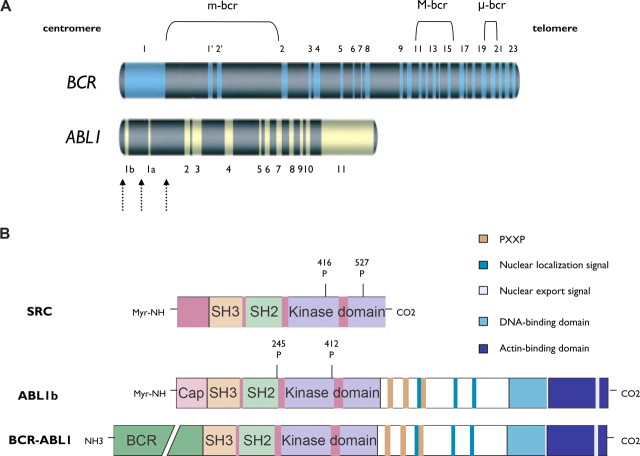
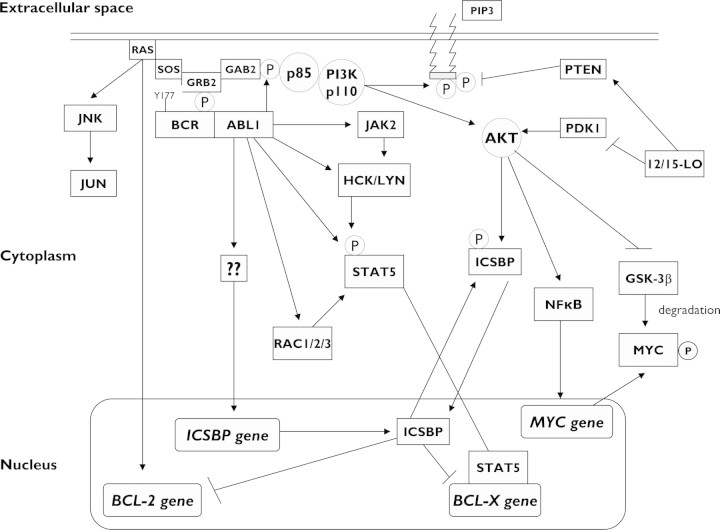
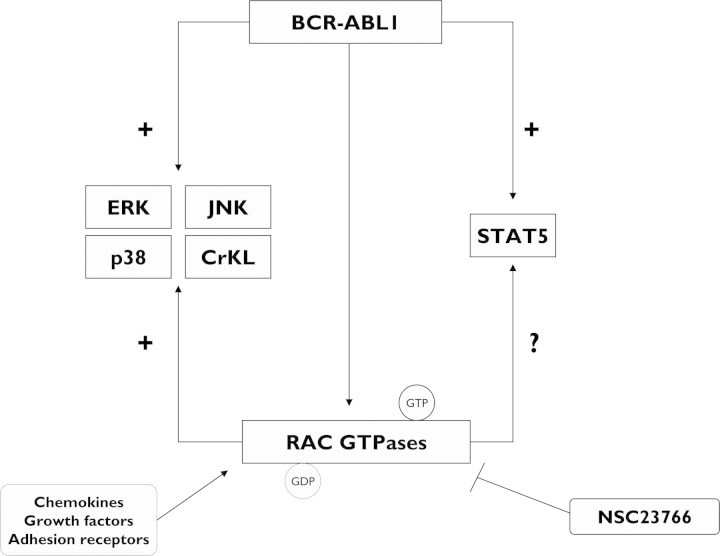
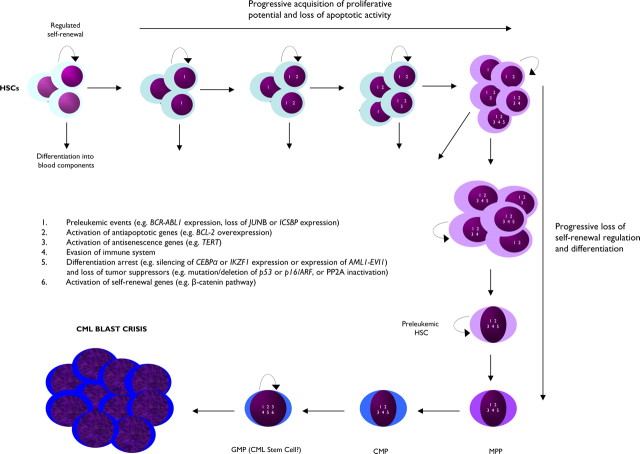
Chronic myeloid leukemia (CML) has been regarded as the paradigmatic example of a malignancy defined by a unique molecular event, the BCR-ABL1 oncogene. Decades of research zeroing in on the role of BCR-ABL1 kinase in the pathogenesis of CML have culminated in the development of highly efficacious therapeutics that, like imatinib mesylate, target the oncogenic kinase activity of BCR-ABL1. In recent years, most research efforts in CML have been devoted to developing novel tyrosine kinase inhibitors (TKIs) as well as to elucidating the mechanisms of resistance to imatinib and other TKIs. Nonetheless, primordial aspects of the pathogenesis of CML, such as the mechanisms responsible for the transition from chronic phase to blast crisis, the causes of genomic instability and faulty DNA repair, the phenomenon of stem cell quiescence, the role of tumor suppressors in TKI resistance and CML progression, or the cross-talk between BCR-ABL1 and other oncogenic signaling pathways, still remain poorly understood. Herein, we synthesize the most relevant and current knowledge on such areas of the pathogenesis of CML.
Figures

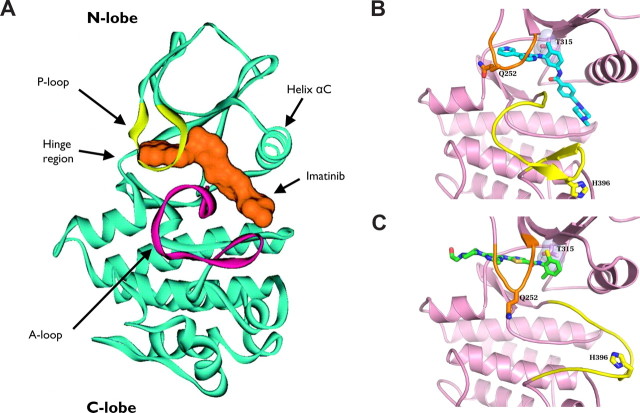
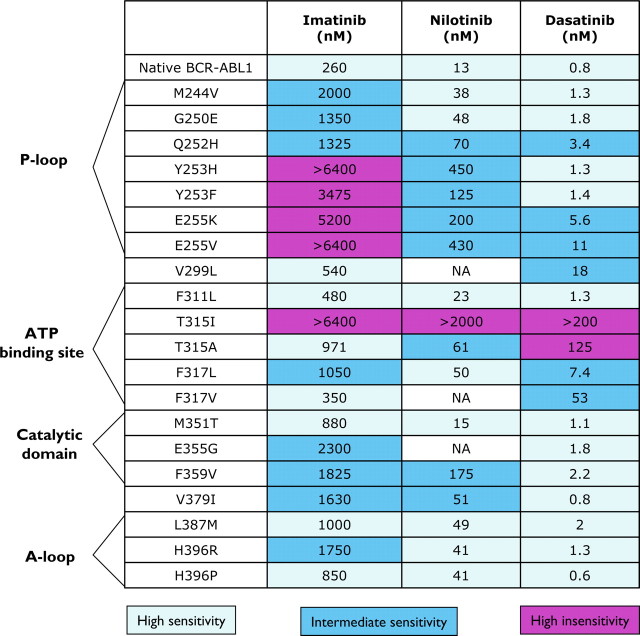
References
-
- Quintás-Cardama A, Cortes JE. Chronic myeloid leukemia: diagnosis and treatment. Mayo Clin Proc. 2006;81:973–988. - PubMed
-
- Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. - PubMed
-
- Ramaraj P, Singh H, Niu N, et al. Effect of mutational inactivation of tyrosine kinase activity on BCR/ABL-induced abnormalities in cell growth and adhesion in human hematopoietic progenitors. Cancer Res. 2004;64:5322–5331. - PubMed
-
- Zhao RC, Jiang Y, Verfaillie CM. A model of human p210(bcr/ABL)-mediated chronic myelogenous leukemia by transduction of primary normal human CD34(+) cells with a BCR/ABL-containing retroviral vector. Blood. 2001;97:2406–2412. - PubMed
-
- Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Acute leukaemia in bcr/abl transgenic mice. Nature. 1990;344:251–253. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
