

Alteration of the mitochondrial apoptotic pathway is key to acquired paclitaxel resistance and can be reversed by ABT-737
- PMID: 18829556
- PMCID: PMC2603173
- DOI: 10.1158/0008-5472.CAN-08-1418
Alteration of the mitochondrial apoptotic pathway is key to acquired paclitaxel resistance and can be reversed by ABT-737
Abstract
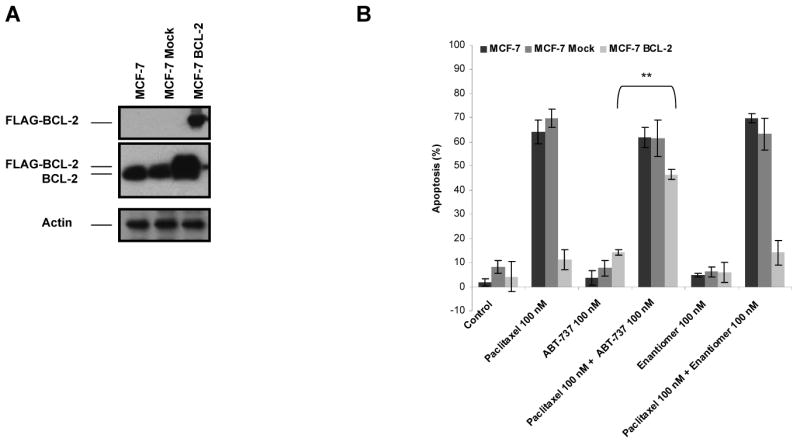
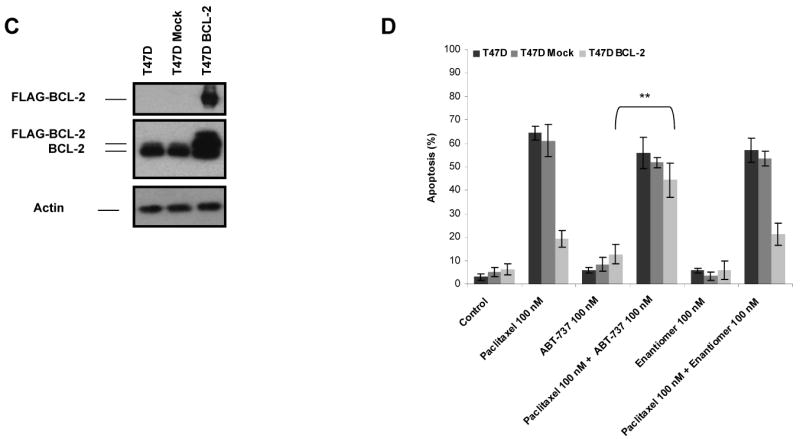
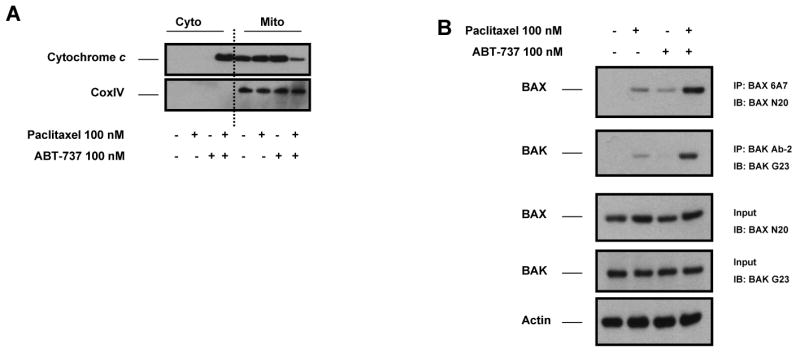
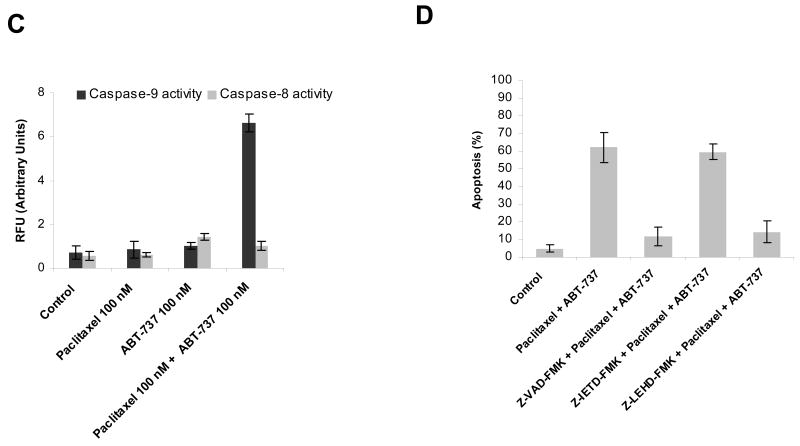
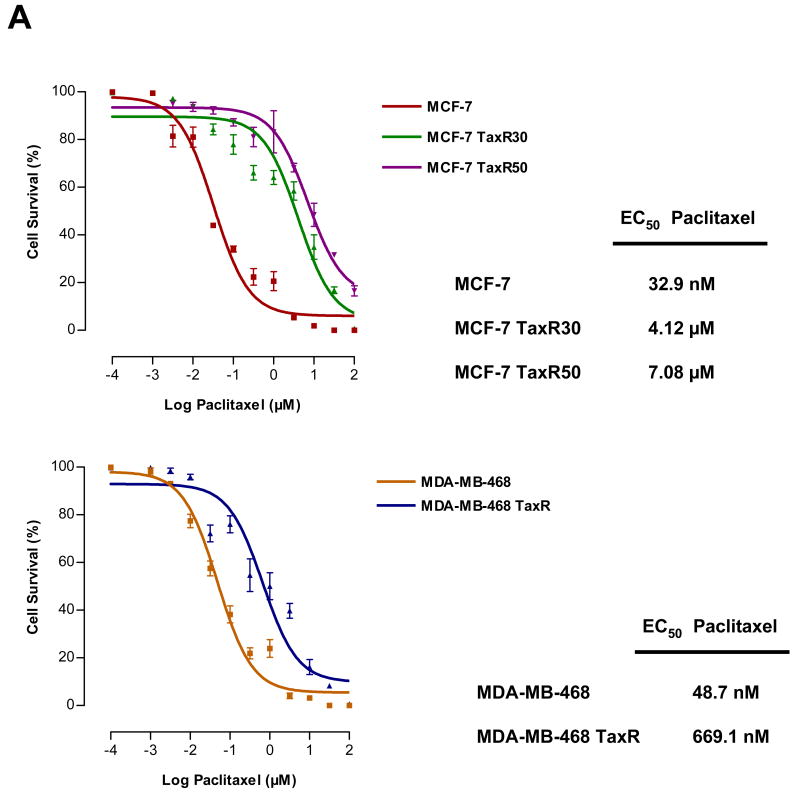
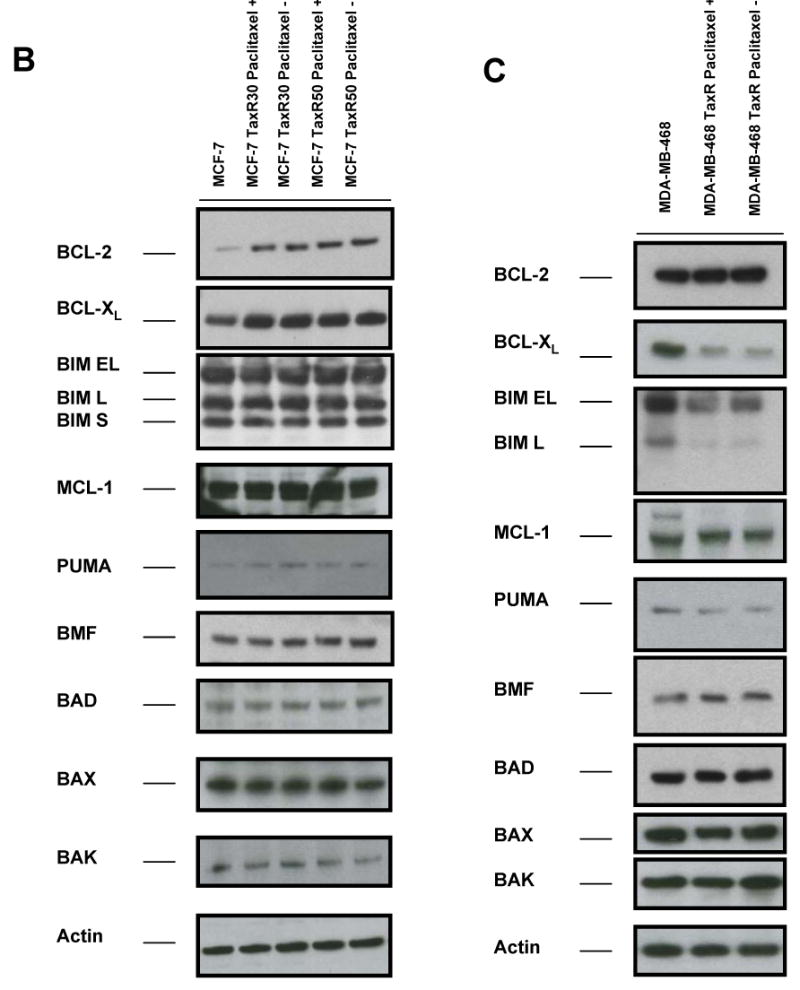
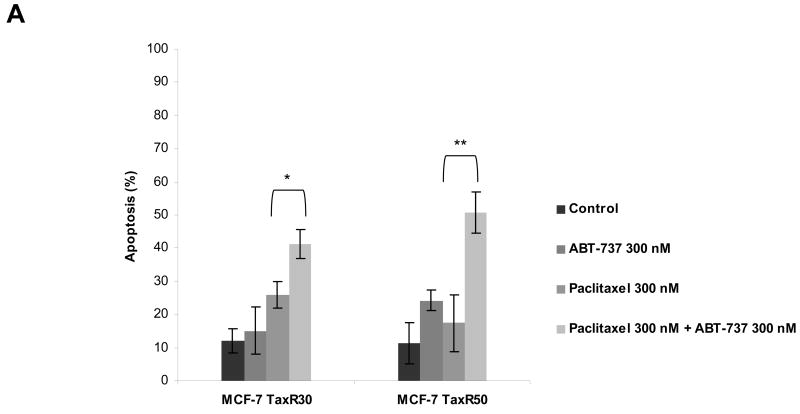
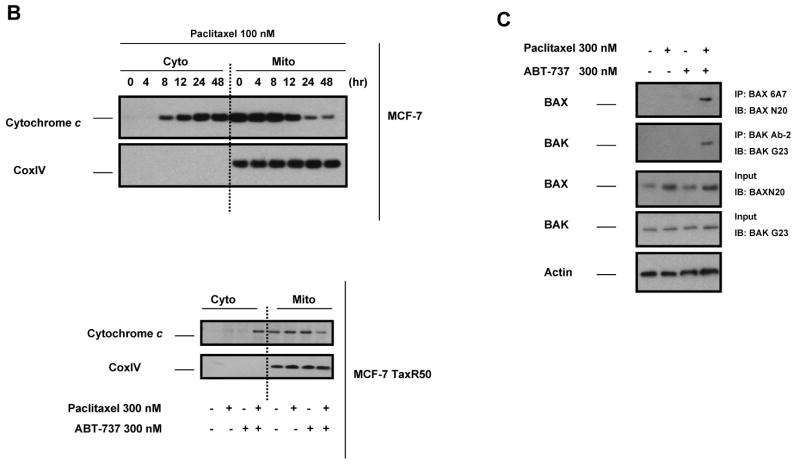
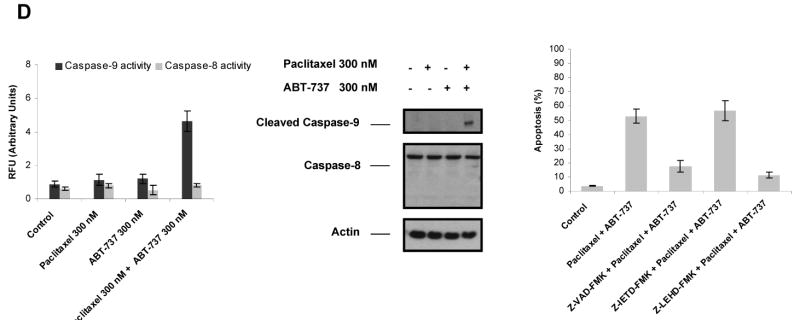
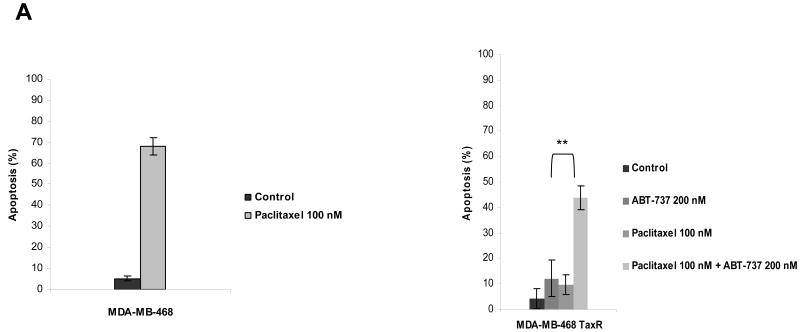
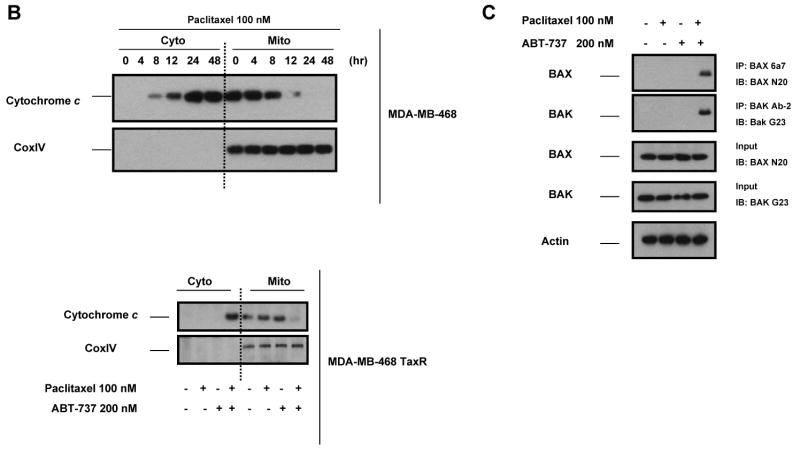
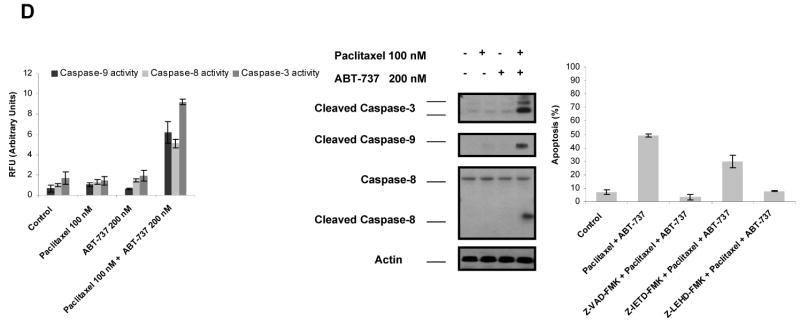
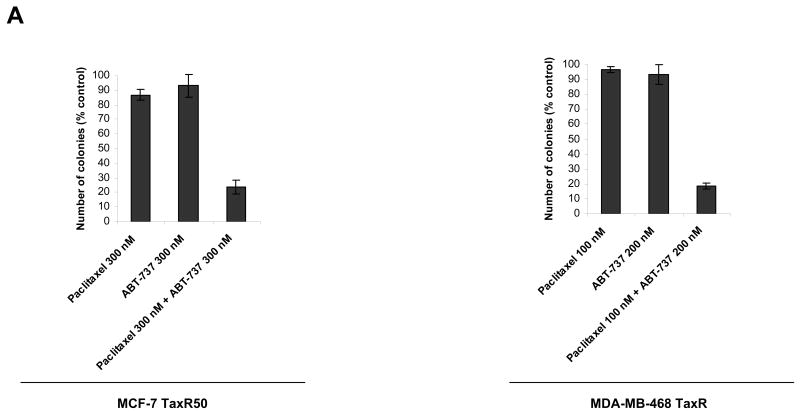
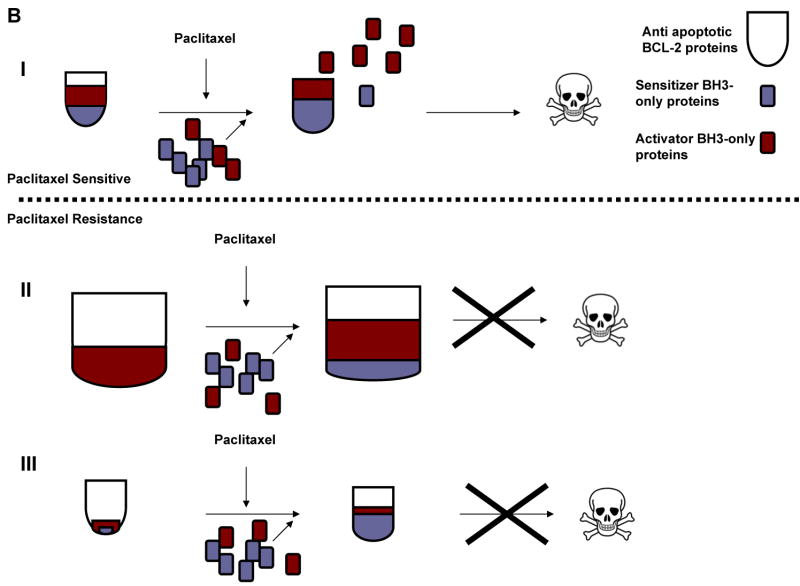
Paclitaxel is a microtubule-targeting antineoplastic drug widely used in human cancers. Even when tumors are initially responsive, progression of disease despite continued taxane therapy is all too common in the treatment of many of the most common epithelial cancers, including breast cancer. However, the mechanisms underlying paclitaxel resistance in cancer cells are not completely understood. Our hypothesis is that changes in the intrinsic (or mitochondrial) cell death pathway controlled by the BCL-2 family are key to the development of acquired paclitaxel resistance. Here we show that paclitaxel activates the mitochondrial apoptosis pathway, which can be blocked by BCL-2 overexpression. Treatment with ABT-737, a small-molecule BCL-2 antagonist, restores sensitivity to paclitaxel in BCL-2-overexpressing cells. To investigate the importance of changes in the intrinsic apoptotic pathway in the absence of enforced BCL-2 expression, we generated two independent breast cancer cell lines with acquired resistance to apoptosis induced by paclitaxel. In these lines, acquired resistance to paclitaxel is mediated either by increased antiapoptotic BCL-2 proteins or decreased proapoptotic BCL-2 proteins. In both cases, ABT-737 can engage the mitochondrial apoptosis pathway to restore sensitivity to paclitaxel to cell lines with acquired paclitaxel resistance. In summary, these findings suggest that alterations in the intrinsic apoptotic pathway controlled by BCL-2 protein family members may be crucial to causing paclitaxel resistance. Furthermore, our results suggest that combining small-molecule BCL-2 antagonists with paclitaxel may offer benefit to patients with paclitaxel-resistant tumors, an oncologic problem of great prevalence.
Figures
References
-
- Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene. 2004;23:2934–49. - PubMed
-
- Crotzer DR, Sun CC, Coleman RL, Wolf JK, Levenback CF, Gershenson DM. Lack of effective systemic therapy for recurrent clear cell carcinoma of the ovary. Gynecol Oncol. 2007;105:404–8. - PubMed
-
- Schrohl AS, Meijer-van Gelder ME, Holten-Andersen MN, et al. Primary tumor levels of tissue inhibitor of metalloproteinases-1 are predictive of resistance to chemotherapy in patients with metastatic breast cancer. Clin Cancer Res. 2006;12:7054–8. - PubMed
-
- Seve P, Isaac S, Tredan O, et al. Expression of class III {beta}-tubulin is predictive of patient outcome in patients with non-small cell lung cancer receiving vinorelbine-based chemotherapy. Clin Cancer Res. 2005;11:5481–6. - PubMed
-
- Wild PJ, Reichle A, Andreesen R, et al. Microsatellite instability predicts poor short-term survival in patients with advanced breast cancer after high-dose chemotherapy and autologous stem-cell transplantation. Clin Cancer Res. 2004;10:556–64. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
