

Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements
- PMID: 18830232
- PMCID: PMC2986177
- DOI: 10.1038/ejhg.2008.178
Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements
Abstract
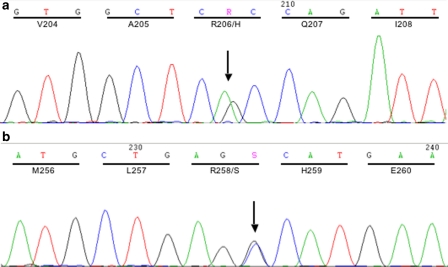
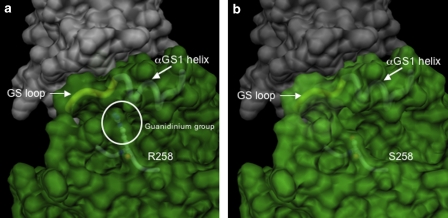
Fibrodysplasia ossificans progressiva (FOP, MIM 135100) is a rare genetic disorder characterized by congenital great toe malformations and progressive heterotopic ossification transforming skeletal muscles and connective tissues to bone following a well-defined anatomic pattern of progression. Recently, FOP has been associated with a specific mutation of ACVR1, the gene coding for a bone morphogenetic protein type I receptor. The identification of ACVR1 as the causative gene for FOP now allows the genetic screening of FOP patients to identify the frequency of the identified recurrent ACVR1 mutation and to investigate genetic variability that may be associated with this severely debilitating disease. We report the screening for mutations in the ACVR1 gene carried out in a cohort of 17 Italian patients. Fifteen of these displayed the previously described c.617G>A mutation, leading to the R206H substitution in the GS domain of the ACVR1 receptor. In two patients, we found a novel mutation c.774G>C, leading to the R258S substitution in the kinase domain of the ACVR1 receptor. In the three-dimensional model of protein structure, R258 maps in close proximity to the GS domain, a key regulator of ACVR1 activity, where R206 is located. The GS domain is known to bind the regulatory protein FKBP12 and to undergo multiple phosphorylation events that trigger a signaling cascade inside the cell. The novel amino-acid substitution is predicted to influence either the conformation/stability of the GS region or the binding affinity with FKBP12, resulting in a less stringent inhibitory control on the ACVR1 kinase activity.
Figures

References
-
- Kaplan FS, Glaser DL, Hebela N, Shore EM. Heterotopic ossification. J Am Acad Orthop Surg. 2004;12:116–125. - PubMed
-
- Kaplan FS, Galser DL, Shore EM, et al. The phenotype of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:201–204.
-
- Pignolo RJ, Suda RK, Kaplan FS. The fibrodysplasia ossificans progressiva lesion. Clin Rev Bone Miner Metab. 2005;3:195–200.
-
- Shore EM, Xu M, Feldman GJ, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–527. - PubMed
-
- Shore EM, Feldman G, Xu M, Kaplan FS. The genetics of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:201–204.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
