

Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease
- PMID: 18830406
- PMCID: PMC2552992
- DOI: 10.1371/journal.pone.0003316
Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease
Abstract
Background: Chronic obstructive pulmonary disease (COPD) is a progressive lung disease characterized by abnormal cellular responses to cigarette smoke, resulting in tissue destruction and airflow limitation. Autophagy is a degradative process involving lysosomal turnover of cellular components, though its role in human diseases remains unclear.
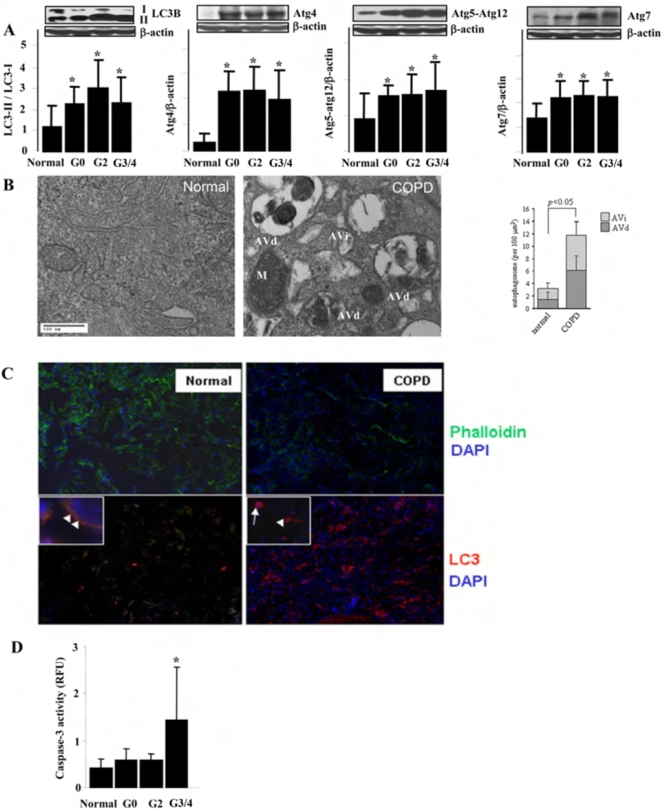
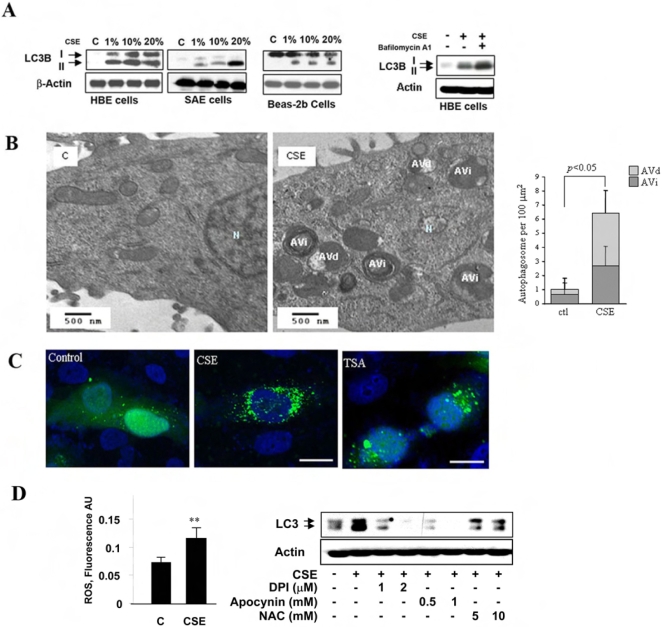
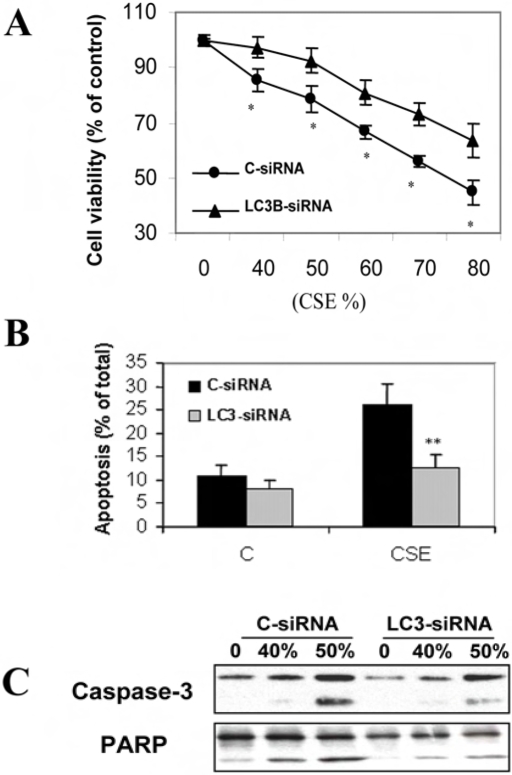
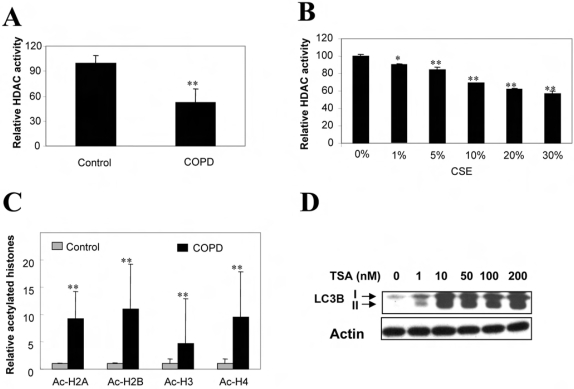
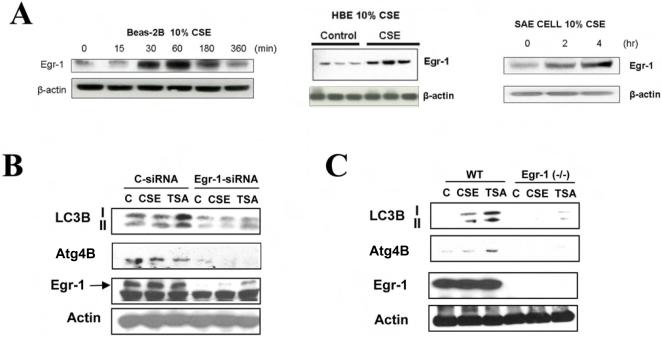
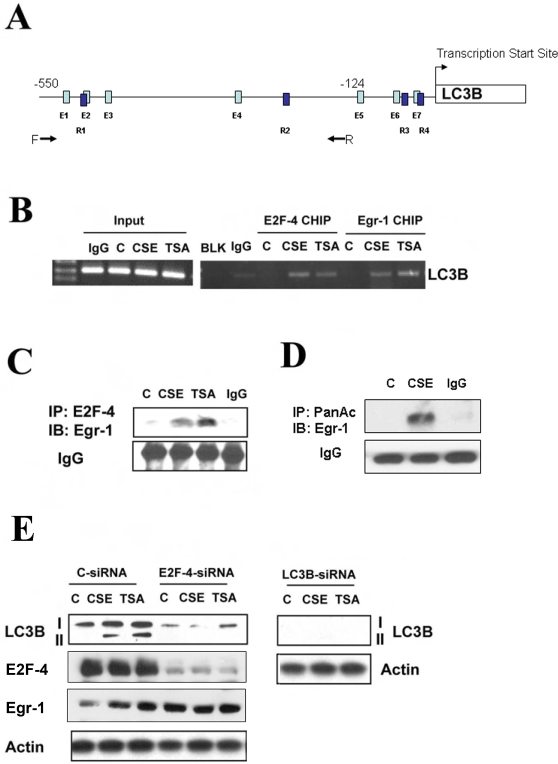
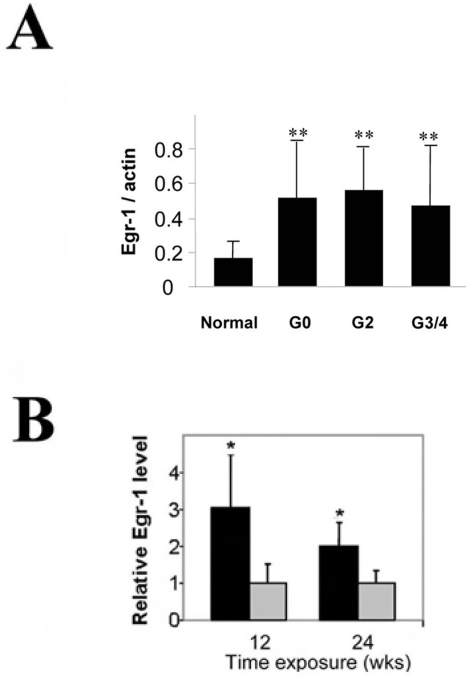
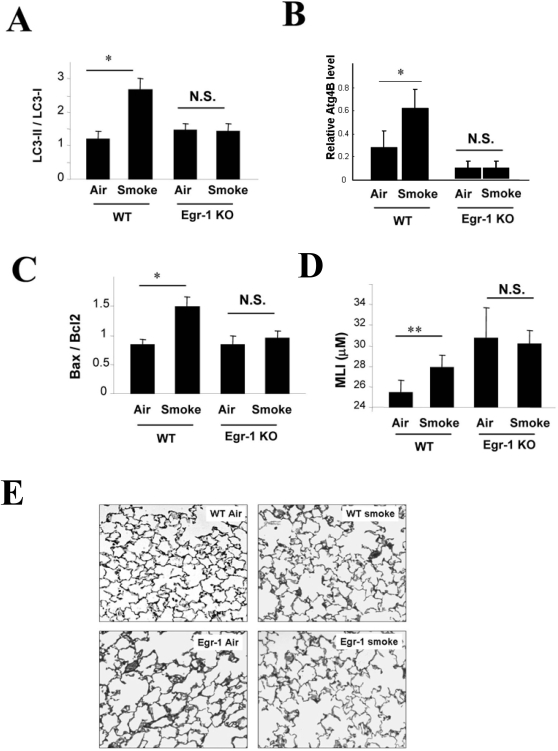
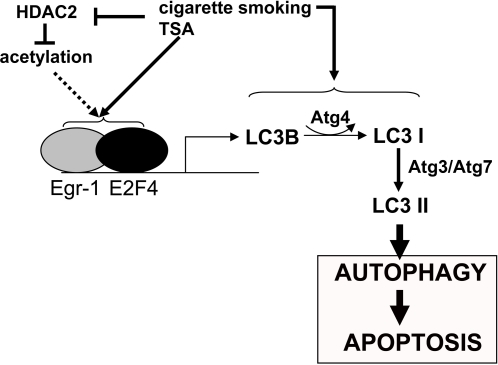
Methodology and principal findings: Increased autophagy was observed in lung tissue from COPD patients, as indicated by electron microscopic analysis, as well as by increased activation of autophagic proteins (microtubule-associated protein-1 light chain-3B, LC3B, Atg4, Atg5/12, Atg7). Cigarette smoke extract (CSE) is an established model for studying the effects of cigarette smoke exposure in vitro. In human pulmonary epithelial cells, exposure to CSE or histone deacetylase (HDAC) inhibitor rapidly induced autophagy. CSE decreased HDAC activity, resulting in increased binding of early growth response-1 (Egr-1) and E2F factors to the autophagy gene LC3B promoter, and increased LC3B expression. Knockdown of E2F-4 or Egr-1 inhibited CSE-induced LC3B expression. Knockdown of Egr-1 also inhibited the expression of Atg4B, a critical factor for LC3B conversion. Inhibition of autophagy by LC3B-knockdown protected epithelial cells from CSE-induced apoptosis. Egr-1(-/-) mice, which displayed basal airspace enlargement, resisted cigarette-smoke induced autophagy, apoptosis, and emphysema.
Conclusions: We demonstrate a critical role for Egr-1 in promoting autophagy and apoptosis in response to cigarette smoke exposure in vitro and in vivo. The induction of autophagy at early stages of COPD progression suggests novel therapeutic targets for the treatment of cigarette smoke induced lung injury.
Conflict of interest statement
Figures
References
-
- Celli BR, MacNee W ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932–946. - PubMed
-
- Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343:269–280. - PubMed
-
- Mayer AS, Newman LS. Genetic and environmental modulation of chronic obstructive pulmonary disease. Respir Physiol. 2001;128:3–11. - PubMed
-
- Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22:672–688. - PubMed
-
- Wang IM, Stepaniants S, Boie Y, Mortimer JR, Kennedy B, et al. Gene expression profiling in patients with chronic obstructive pulmonary disease and lung cancer. Am J Respir Crit Care Med. 2007;177:402–411. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
