

The FEATURE framework for protein function annotation: modeling new functions, improving performance, and extending to novel applications
- PMID: 18831785
- PMCID: PMC2559884
- DOI: 10.1186/1471-2164-9-S2-S2
The FEATURE framework for protein function annotation: modeling new functions, improving performance, and extending to novel applications
Abstract
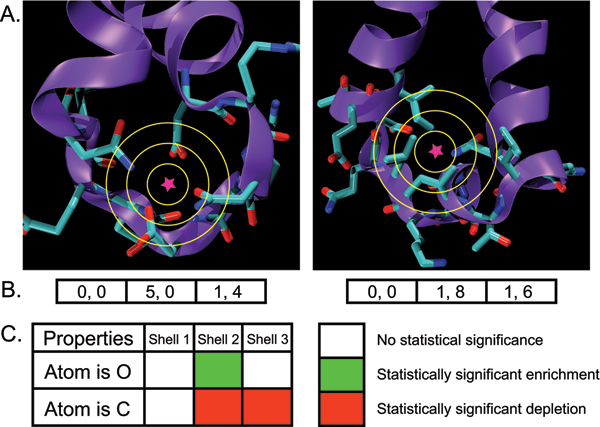
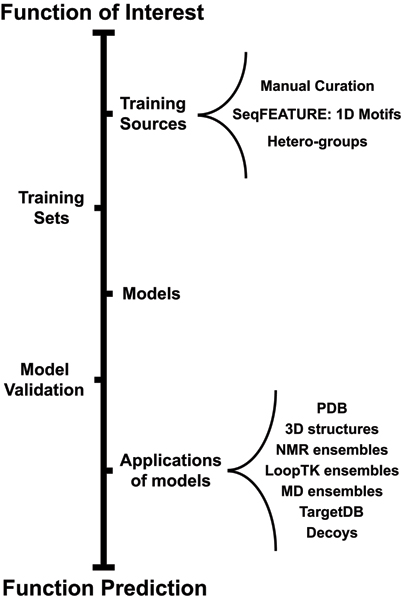
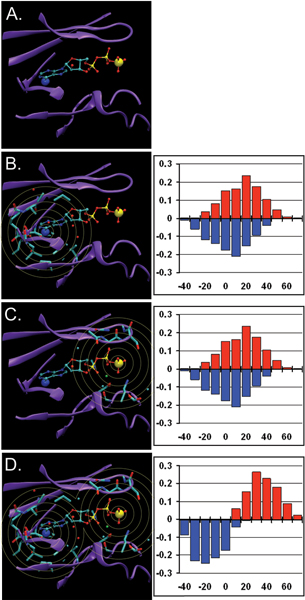
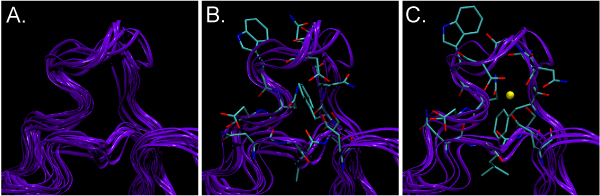
Structural genomics efforts contribute new protein structures that often lack significant sequence and fold similarity to known proteins. Traditional sequence and structure-based methods may not be sufficient to annotate the molecular functions of these structures. Techniques that combine structural and functional modeling can be valuable for functional annotation. FEATURE is a flexible framework for modeling and recognition of functional sites in macromolecular structures. Here, we present an overview of the main components of the FEATURE framework, and describe the recent developments in its use. These include automating training sets selection to increase functional coverage, coupling FEATURE to structural diversity generating methods such as molecular dynamics simulations and loop modeling methods to improve performance, and using FEATURE in large-scale modeling and structure determination efforts.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
