

Structure of the human voltage-dependent anion channel
- PMID: 18832158
- PMCID: PMC2557026
- DOI: 10.1073/pnas.0808115105
Structure of the human voltage-dependent anion channel
Abstract
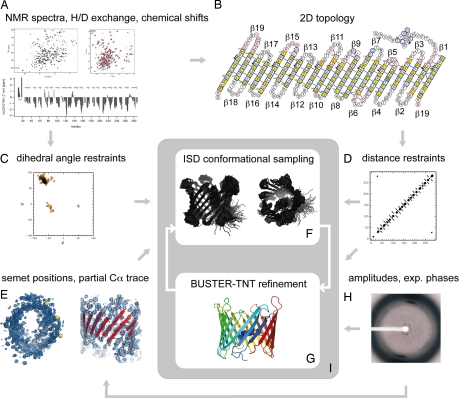
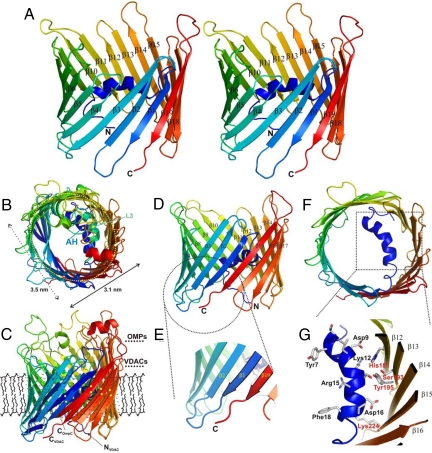
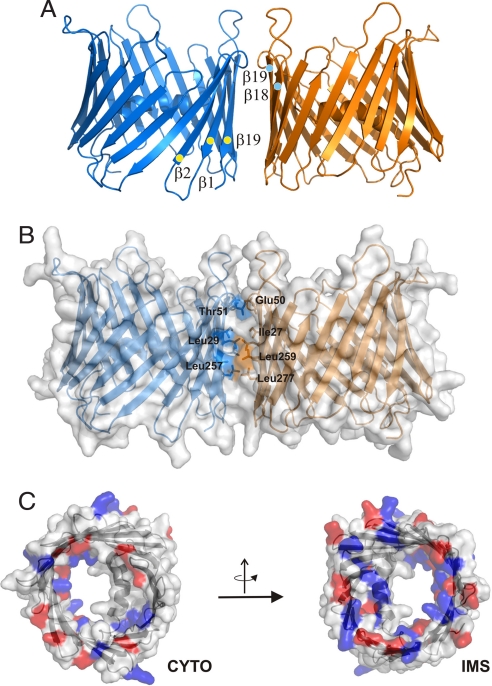
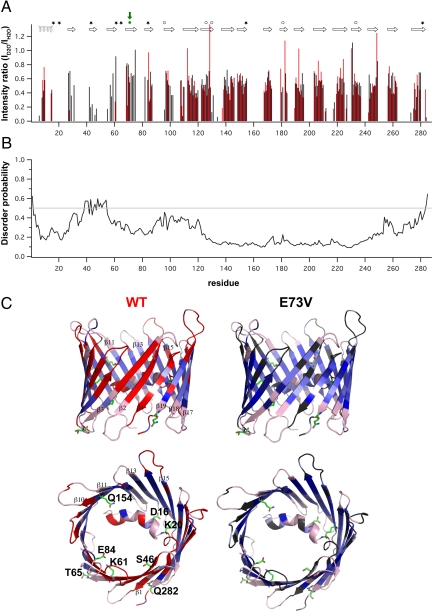
The voltage-dependent anion channel (VDAC), also known as mitochondrial porin, is the most abundant protein in the mitochondrial outer membrane (MOM). VDAC is the channel known to guide the metabolic flux across the MOM and plays a key role in mitochondrially induced apoptosis. Here, we present the 3D structure of human VDAC1, which was solved conjointly by NMR spectroscopy and x-ray crystallography. Human VDAC1 (hVDAC1) adopts a beta-barrel architecture composed of 19 beta-strands with an alpha-helix located horizontally midway within the pore. Bioinformatic analysis indicates that this channel architecture is common to all VDAC proteins and is adopted by the general import pore TOM40 of mammals, which is also located in the MOM.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–749. - PubMed
-
- Hill K, et al. Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins [see comment] Nature. 1998;395:516–521. - PubMed
-
- Wiedemann N, et al. Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature. 2003;424:565–571. - PubMed
-
- Schein SJ, Colombini M, Finkelstein A. Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J Membr Biol. 1976;30:99–120. - PubMed
-
- Blachly-Dyson E, Forte M. VDAC channels. IUBMB Life. 2001;52:113–118. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
