

Modeling genetic inheritance of copy number variations
- PMID: 18832372
- PMCID: PMC2588508
- DOI: 10.1093/nar/gkn641
Modeling genetic inheritance of copy number variations
Abstract
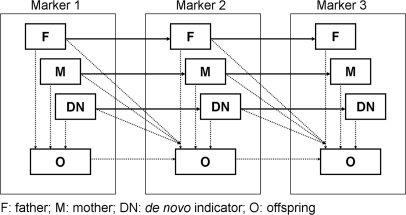
Copy number variations (CNVs) are being used as genetic markers or functional candidates in gene-mapping studies. However, unlike single nucleotide polymorphism or microsatellite genotyping techniques, most CNV detection methods are limited to detecting total copy numbers, rather than copy number in each of the two homologous chromosomes. To address this issue, we developed a statistical framework for intensity-based CNV detection platforms using family data. Our algorithm identifies CNVs for a family simultaneously, thus avoiding the generation of calls with Mendelian inconsistency while maintaining the ability to detect de novo CNVs. Applications to simulated data and real data indicate that our method significantly improves both call rates and accuracy of boundary inference, compared to existing approaches. We further illustrate the use of Mendelian inheritance to infer SNP allele compositions in each of the two homologous chromosomes in CNV regions using real data. Finally, we applied our method to a set of families genotyped using both the Illumina HumanHap550 and Affymetrix genome-wide 5.0 arrays to demonstrate its performance on both inherited and de novo CNVs. In conclusion, our method produces accurate CNV calls, gives probabilistic estimates of CNV transmission and builds a solid foundation for the development of linkage and association tests utilizing CNVs.
Figures




References
-
- Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat. Rev. Genet. 2006;7:85–97. - PubMed
-
- Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat. Rev. Genet. 2007;8:639–646. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
