

Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia
- PMID: 18832465
- PMCID: PMC2572931
- DOI: 10.1073/pnas.0808084105
Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia
Abstract
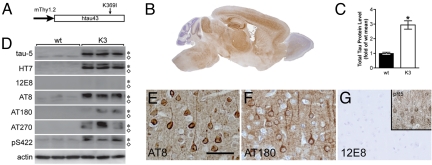
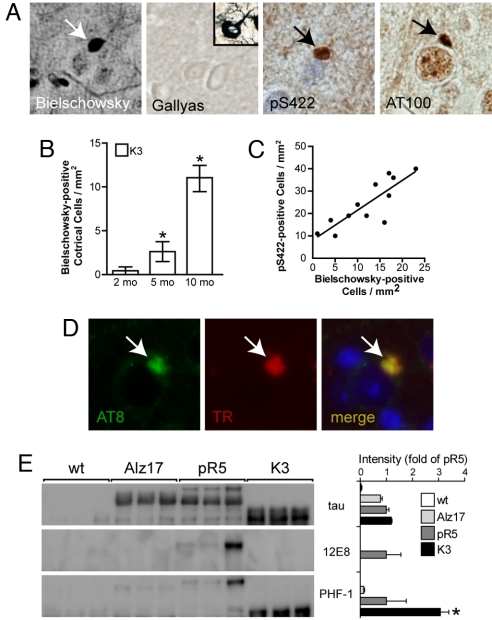
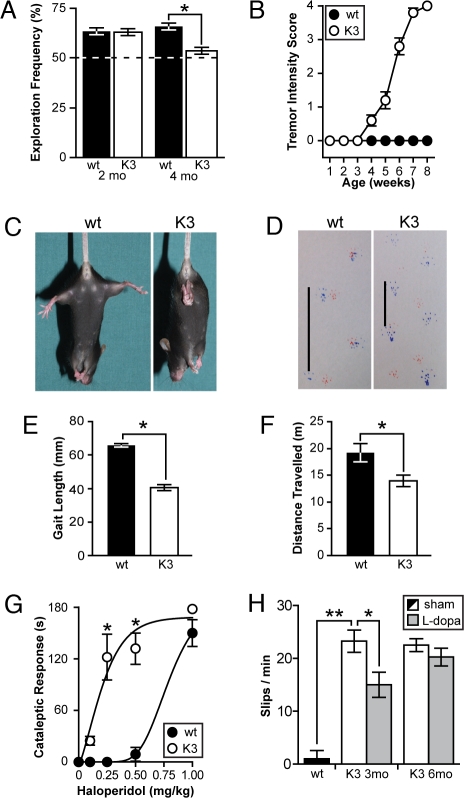
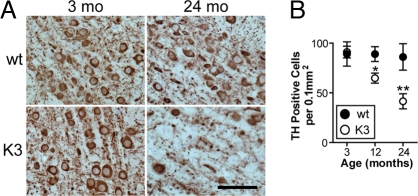
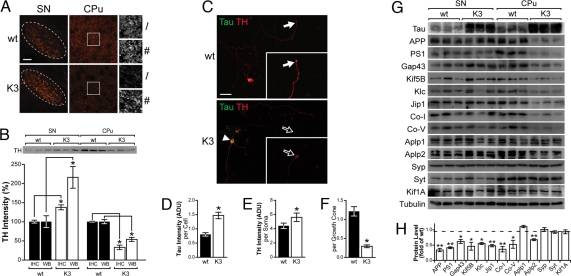
Frontotemporal dementia (FTD) is characterized by cognitive and behavioral changes and, in a significant subset of patients, Parkinsonism. Histopathologically, FTD frequently presents with tau-containing lesions, which in familial cases result from mutations in the MAPT gene encoding tau. Here we present a novel transgenic mouse strain (K3) that expresses human tau carrying the FTD mutation K369I. K3 mice develop a progressive histopathology that is reminiscent of that in human FTD with the K369I mutation. In addition, K3 mice show early-onset memory impairment and amyotrophy in the absence of overt neurodegeneration. Different from our previously generated tau transgenic strains, the K3 mice express the transgene in the substantia nigra (SN) and show an early-onset motor phenotype that reproduces Parkinsonism with tremor, bradykinesia, abnormal gait, and postural instability. Interestingly, motor performance of young, but not old, K3 mice improves upon L-dopa treatment, which bears similarities to Parkinsonism in FTD. The early-onset symptoms in the K3 mice are mechanistically related to selectively impaired anterograde axonal transport of distinct cargos, which precedes the loss of dopaminergic SN neurons that occurs in aged mice. The impaired axonal transport in SN neurons affects, among others, vesicles containing the dopamine-synthesizing enzyme tyrosine hydroxylase. Distinct modes of transport are also impaired in sciatic nerves, which may explain amyotrophy. Together, the K3 mice are a unique model of FTD-associated Parkinsonism, with pathomechanistic implications for the human pathologic process.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. - PubMed
-
- Baker M, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. - PubMed
-
- Cruts M, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. - PubMed
-
- Gotz J, Ittner LM. Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci. 2008;9:532–544. - PubMed
-
- Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
