

The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry
- PMID: 18833199
- PMCID: PMC2728452
- DOI: 10.1038/nprot.2008.150
The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry
Abstract
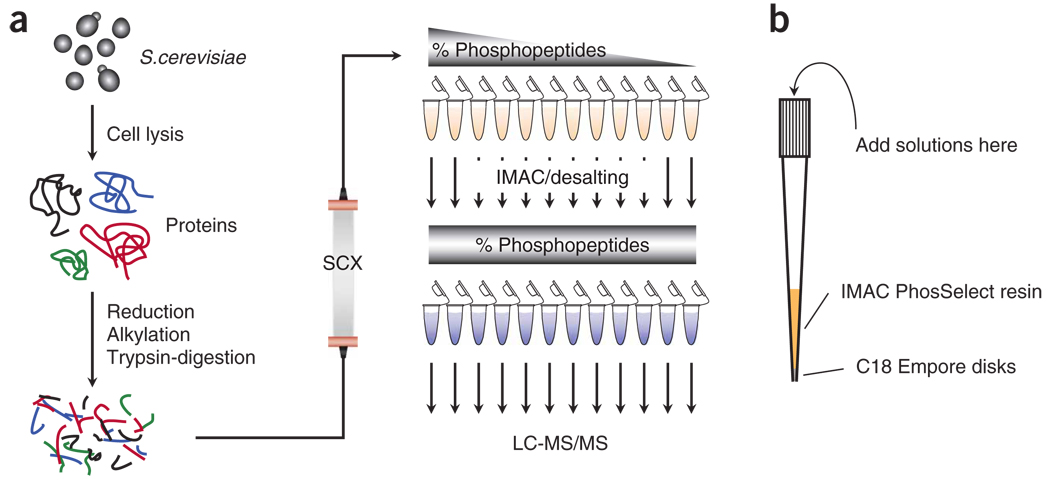
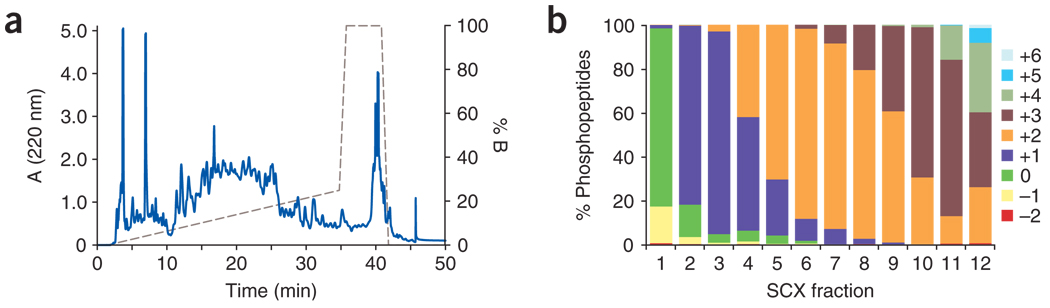
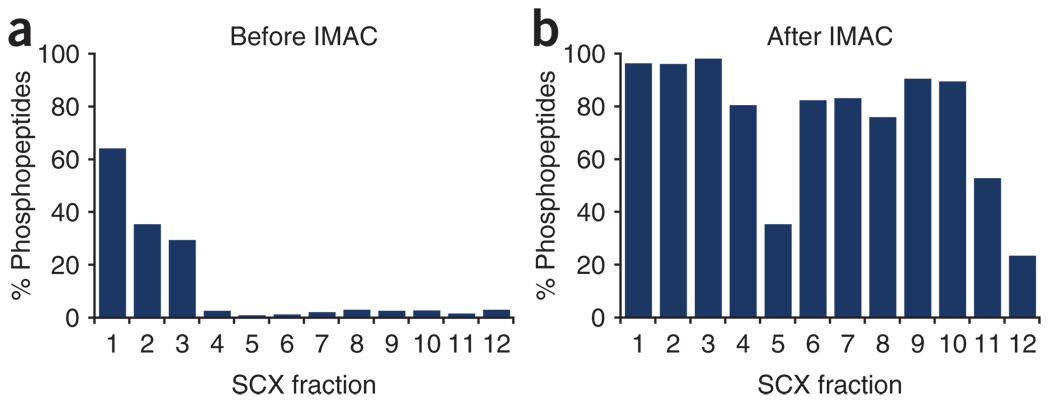
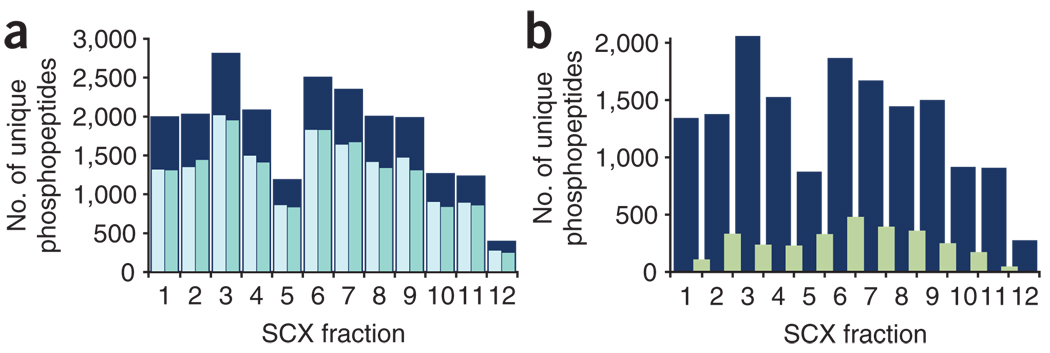
The success in profiling the phosphoproteome by mass spectrometry-based proteomics has been intimately related to the availability of methods that selectively enrich for phosphopeptides. To this end, we describe a protocol that combines two sequential enrichment steps. First, strong cation exchange (SCX) chromatography separates peptides by solution charge. Phosphate groups contribute to solution charge by adding a negative charge at pH 2.7. Therefore, at that pH, phosphopeptides are expected to elute earlier than their nonphosphorylated homologs. Second, immobilized metal affinity chromatography (IMAC) takes advantage of phosphate's affinity for metal ions such as Fe(3+) to uniformly enrich for phosphopeptides from the previously collected SCX fractions. We have successfully employed the SCX/IMAC enrichment strategy in the exploration of phosphoproteomes from several systems including mouse liver and Drosophila embryos characterizing over 5,500 and 13,000 phosphorylation events, respectively. The SCX/IMAC enrichment protocol requires 2 days, and the entire procedure from cells to a phosphorylation data set can be completed in less than 10 days.
Figures
References
-
- Hastie CJ, McLauchlan HJ, Cohen P. Assay of protein kinases using radiolabeled ATP: a protocol. Nat. Protoc. 2006;1:968–971. - PubMed
-
- Collins MO, Yu L, Choudhary JS. Analysis of protein phosphorylation on a proteome-scale. Proteomics. 2007;7:2751–2768. - PubMed
-
- Hunter T, Cooper JA. Protein-tyrosine kinases. Annu. Rev. Biochem. 1985;54:897–930. - PubMed
-
- Rush J, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 2005;23:94–101. - PubMed
-
- Andersson L, Porath J. Isolation of phosphoproteins by immobilized metal (Fe3+) affinity chromatography. Anal. Biochem. 1986;154:250–254. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
